

Identification of SCN1A and PCDH19 mutations in Chinese children with Dravet syndrome
- PMID: 22848613
- PMCID: PMC3405017
- DOI: 10.1371/journal.pone.0041802
Identification of SCN1A and PCDH19 mutations in Chinese children with Dravet syndrome
Abstract
Background: Dravet syndrome is a severe form of epilepsy. Majority of patients have a mutation in SCN1A gene, which encodes a voltage-gated sodium channel. A recent study has demonstrated that 16% of SCN1A-negative patients have a mutation in PCDH19, the gene encoding protocadherin-19. Mutations in other genes account for only a very small proportion of families. TSPYL4 is a novel candidate gene within the locus 6q16.3-q22.31 identified by linkage study.
Objective: The present study examined the mutations in epileptic Chinese children with emphasis on Dravet syndrome.
Methods: A hundred children with severe epilepsy were divided into Dravet syndrome and non-Dravet syndrome groups and screened for SCN1A mutations by direct sequencing. SCN1A-negative Dravet syndrome patients and patients with phenotypes resembling Dravet syndrome were checked for PCDH19 and TSPYL4 mutations.
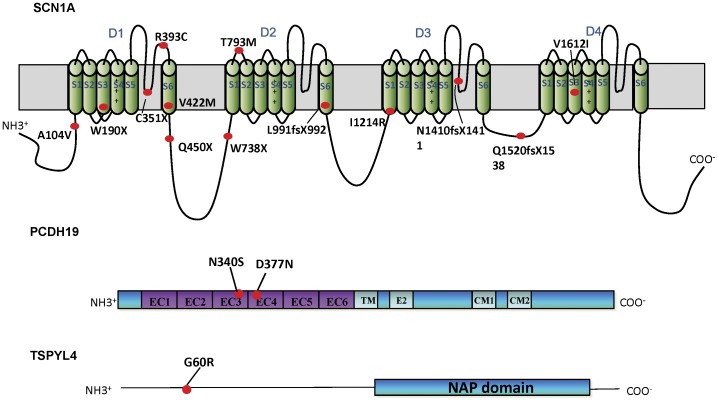
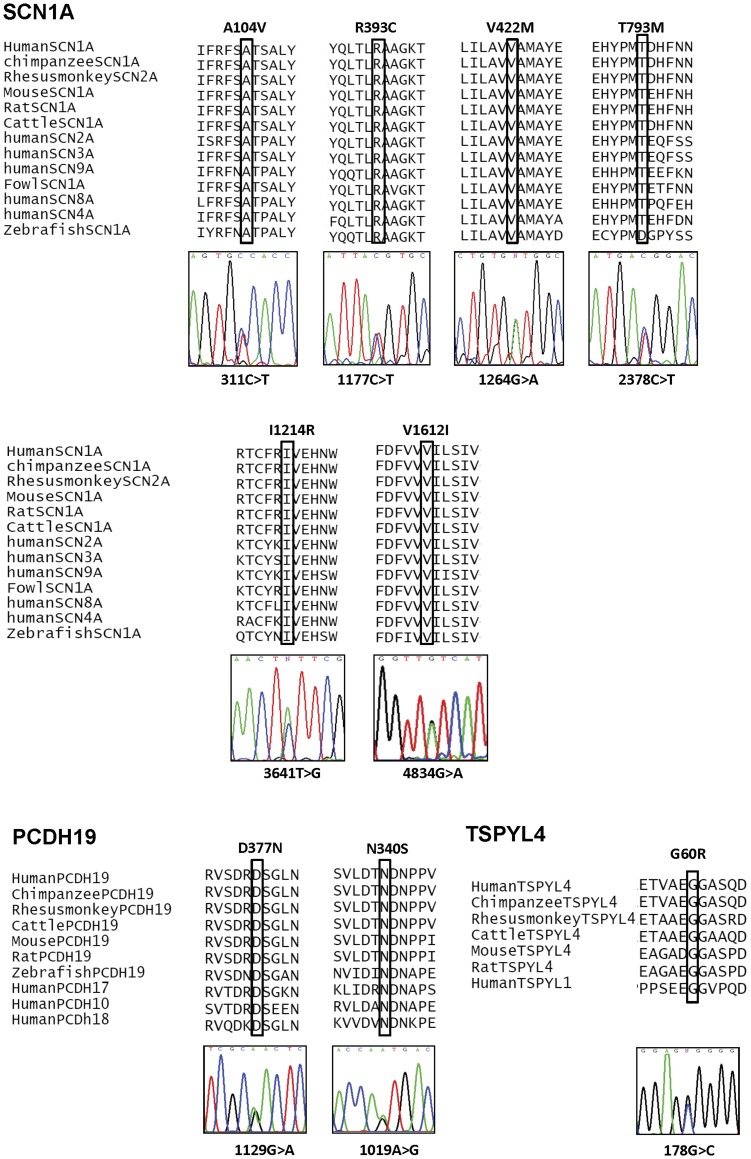
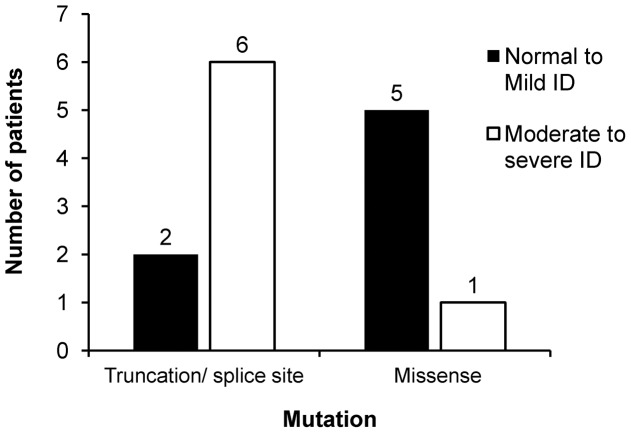
Results: Eighteen patients (9 males, 9 females) were diagnosed to have Dravet syndrome. Among them, 83% (15/18) had SCN1A mutations including truncating (7), splice site (2) and missense mutations (6). The truncating/splice site mutations were associated with moderate to severe degree of intellectual disability (p<0.05). During the progression of disease, 73% (11/15) had features fitting into the diagnostic criteria of autism spectrum disorder and 53% (8/15) had history of vaccination-induced seizures. A novel PCDH19 p.D377N mutation was identified in one SCN1A-negative female patient with Dravet syndrome and a known PCDH19 p.N340S mutation in a female non-Dravet syndrome patient. The former also inherited a TSPYL4 p.G60R variant.
Conclusion: A high percentage of SCN1A mutations was identified in our Chinese cohort of Dravet syndrome patients but none in the rest of patients. We demonstrated that truncating/splice site mutations were linked to moderate to severe intellectual disability in these patients. A de novo PCDH19 missense mutation together with an inherited TSPYL4 missense variant were identified in a patient with Dravet syndrome.
Conflict of interest statement
Figures
Similar articles
-
Clinical and molecular analysis of epilepsy-related genes in patients with Dravet syndrome.Medicine (Baltimore). 2018 Dec;97(50):e13565. doi: 10.1097/MD.0000000000013565. Medicine (Baltimore). 2018. PMID: 30558019 Free PMC article.
-
Dravet syndrome and Dravet syndrome-like phenotype: a systematic review of the SCN1A and PCDH19 variants.Neurogenetics. 2021 May;22(2):105-115. doi: 10.1007/s10048-021-00644-7. Epub 2021 May 3. Neurogenetics. 2021. PMID: 33937968
-
Genetics and clinical correlation of Dravet syndrome and its mimics - experience of a tertiary center in Taiwan.Pediatr Neonatol. 2021 Sep;62(5):550-558. doi: 10.1016/j.pedneo.2021.05.022. Epub 2021 Jun 23. Pediatr Neonatol. 2021. PMID: 34226156
-
Clinical implications of SCN1A missense and truncation variants in a large Japanese cohort with Dravet syndrome.Epilepsia. 2017 Feb;58(2):282-290. doi: 10.1111/epi.13639. Epub 2016 Dec 24. Epilepsia. 2017. PMID: 28012175
-
Dravet syndrome as part of the clinical and genetic spectrum of sodium channel epilepsies and encephalopathies.Epilepsia. 2019 Dec;60 Suppl 3:S2-S7. doi: 10.1111/epi.16054. Epilepsia. 2019. PMID: 31904125 Review.
Cited by
-
Clinical and genetic aspects of PCDH19-related epilepsy syndromes and the possible role of PCDH19 mutations in males with autism spectrum disorders.Neurogenetics. 2013 Feb;14(1):23-34. doi: 10.1007/s10048-013-0353-1. Epub 2013 Jan 20. Neurogenetics. 2013. PMID: 23334464
-
The Broad Clinical Spectrum of Epilepsies Associated With Protocadherin 19 Gene Mutation.Front Neurol. 2022 Jan 17;12:780053. doi: 10.3389/fneur.2021.780053. eCollection 2021. Front Neurol. 2022. PMID: 35111125 Free PMC article. Review.
-
Moving gating charges through the gating pore in a Kv channel voltage sensor.Proc Natl Acad Sci U S A. 2014 May 13;111(19):E1950-9. doi: 10.1073/pnas.1406161111. Epub 2014 Apr 29. Proc Natl Acad Sci U S A. 2014. PMID: 24782544 Free PMC article.
-
SCN1A Gene Mutation and Adaptive Functioning in 18 Vietnamese Children with Dravet Syndrome.J Clin Neurol. 2017 Jan;13(1):62-70. doi: 10.3988/jcn.2017.13.1.62. J Clin Neurol. 2017. PMID: 28079314 Free PMC article.
-
A rise in saliva and urine pH in children with SCN1A-related epilepsy: An exploratory prospective controlled study.Front Neurol. 2022 Sep 27;13:982050. doi: 10.3389/fneur.2022.982050. eCollection 2022. Front Neurol. 2022. PMID: 36237607 Free PMC article.
References
-
- Audenaert D, Van Broeckhoven C, De Jonghe P. Genes and loci involved in febrile seizures and related epilepsy syndromes. Hum Mutat. 2006;27:391–401. - PubMed
-
- Singh R, Andermann E, Whitehouse WP, Harvey AS, Keene DL, et al. Severe myoclonic epilepsy of infancy: extended spectrum of GEFS+? Epilepsia. 2001;42:837–844. - PubMed
-
- Dravet C. The core Dravet syndrome phenotype. Epilepsia. 2011;52:3–9. - PubMed
-
- Dravet C, Bureau M, Oguni H, Fukuyama Y, Cokar O. Roger JB, Dravet C, Genton P, Tassinari C, Wolf P, editors. Severe myoclonic epilepsy in infancy (Dravet syndrome). 2005. pp. 89–113. editor. Epileptic Syndromes in Infancy, Childhood and Adolescence. London: John Libbey. - PubMed
-
- Harkin LA, McMahon JM, Iona X, Dibbens L, Pelekanos JT, et al. The spectrum of SCN1A-related infantile epileptic encephalopathies. Brain. 2007;130:843–852. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
