

Dose-dependent effects of small-molecule antagonists on the genomic landscape of androgen receptor binding
- PMID: 22849360
- PMCID: PMC3507642
- DOI: 10.1186/1471-2164-13-355
Dose-dependent effects of small-molecule antagonists on the genomic landscape of androgen receptor binding
Abstract
Background: The androgen receptor plays a critical role throughout the progression of prostate cancer and is an important drug target for this disease. While chromatin immunoprecipitation coupled with massively parallel sequencing (ChIP-Seq) is becoming an essential tool for studying transcription and chromatin modification factors, it has rarely been employed in the context of drug discovery.
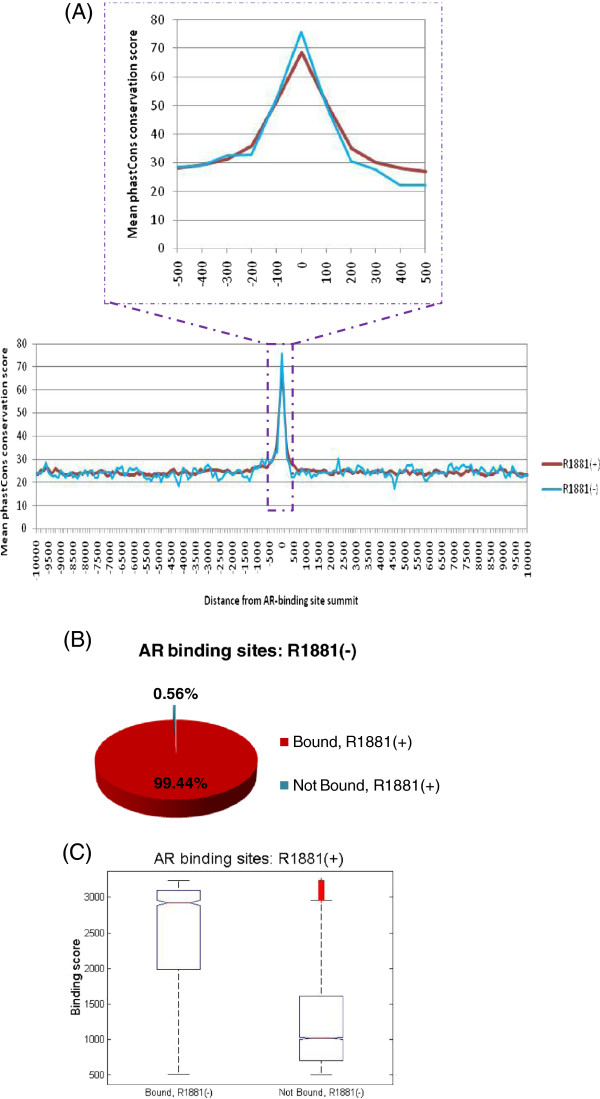
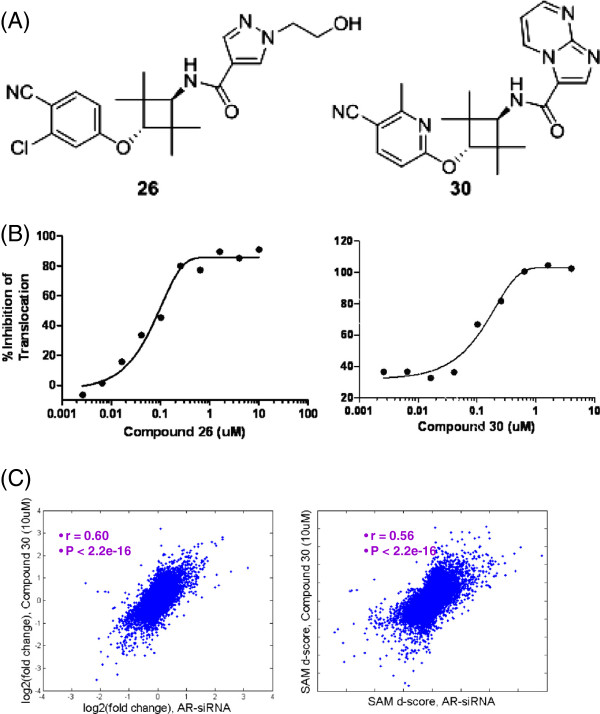
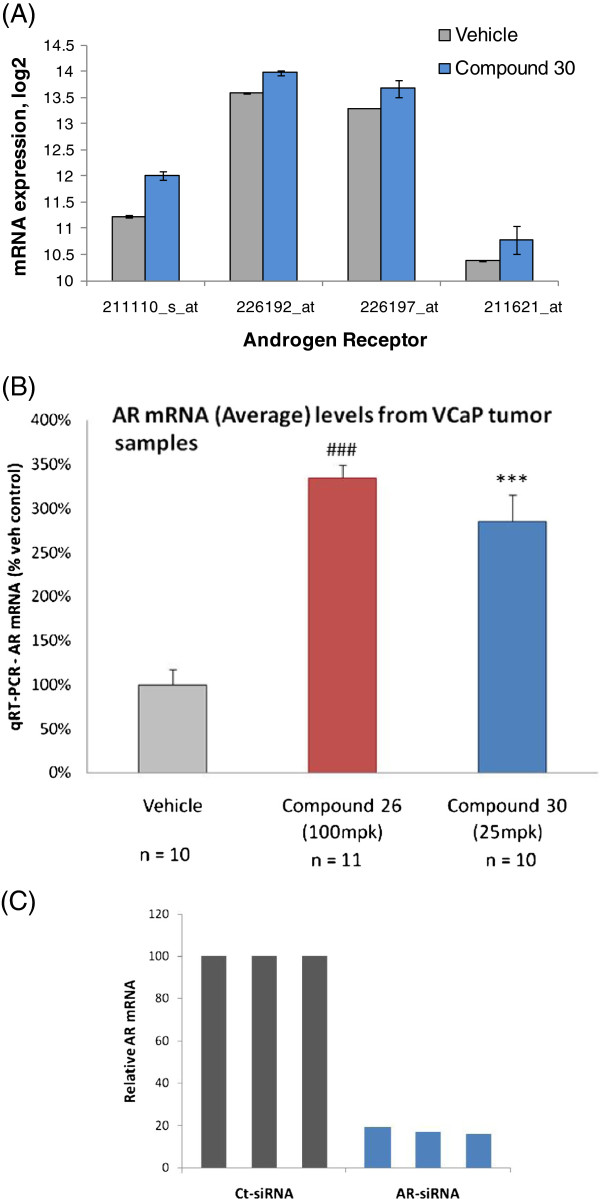
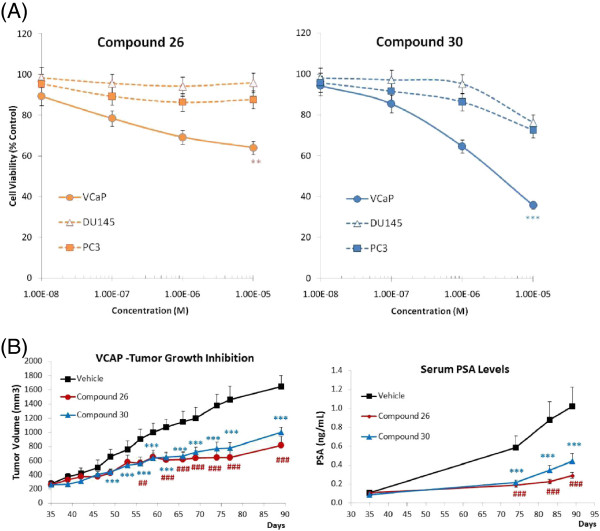
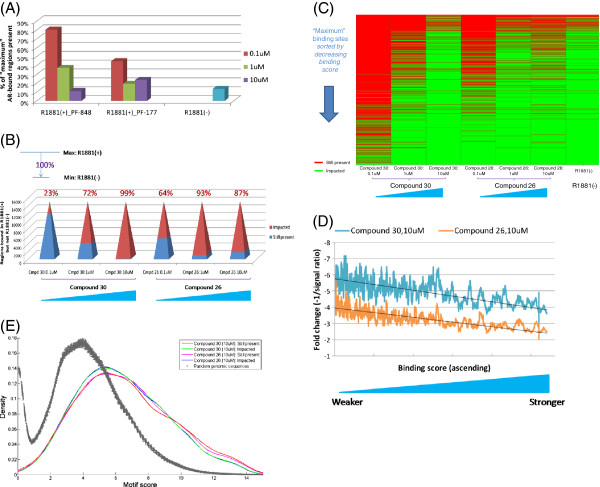
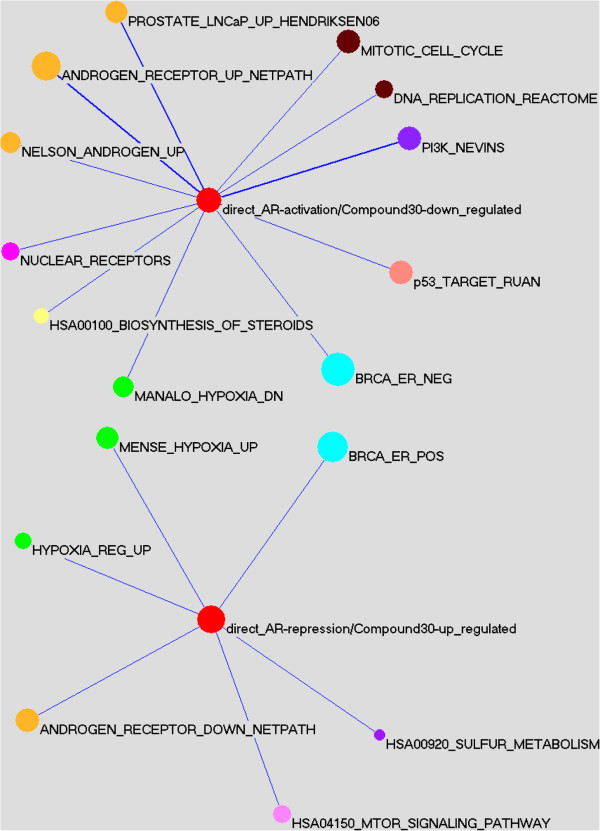
Results: Here we report changes in the genome-wide AR binding landscape due to dose-dependent inhibition by drug-like small molecules using ChIP-Seq. Integration of sequence analysis, transcriptome profiling, cell viability assays and xenograft tumor growth inhibition studies enabled us to establish a direct cistrome-activity relationship for two novel potent AR antagonists. By selectively occupying the strongest binding sites, AR signaling remains active even when androgen levels are low, as is characteristic of first-line androgen ablation therapy. Coupled cistrome and transcriptome profiling upon small molecule antagonism led to the identification of a core set of AR direct effector genes that are most likely to mediate the activities of targeted agents: unbiased pathway mapping revealed that AR is a key modulator of steroid metabolism by forming a tightly controlled feedback loop with other nuclear receptor family members and this oncogenic effect can be relieved by antagonist treatment. Furthermore, we found that AR also has an extensive role in negative gene regulation, with estrogen (related) receptor likely mediating its function as a transcriptional repressor.
Conclusions: Our study provides a global and dynamic view of AR's regulatory program upon antagonism, which may serve as a molecular basis for deciphering and developing AR therapeutics.
Figures
References
-
- Koivisto P, Kononen J, Palmberg C, Tammela T, Hyytinen E, Isola J, Trapman J, Cleutjens K, Noordzij A, Visakorpi T. K: Androgen receptor gene amplification: a possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. Cancer Res. 1997;57:314–319. - PubMed
-
- Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinänen R, Palmberg C, Palotie A, Tammela T, Isola J. K: vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet. 1995;4:401–406. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
