

A maximum-likelihood method to correct for allelic dropout in microsatellite data with no replicate genotypes
- PMID: 22851645
- PMCID: PMC3660999
- DOI: 10.1534/genetics.112.139519
A maximum-likelihood method to correct for allelic dropout in microsatellite data with no replicate genotypes
Abstract
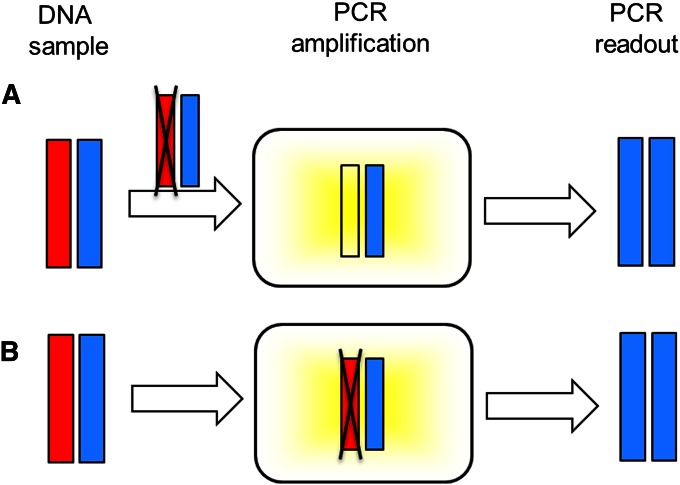
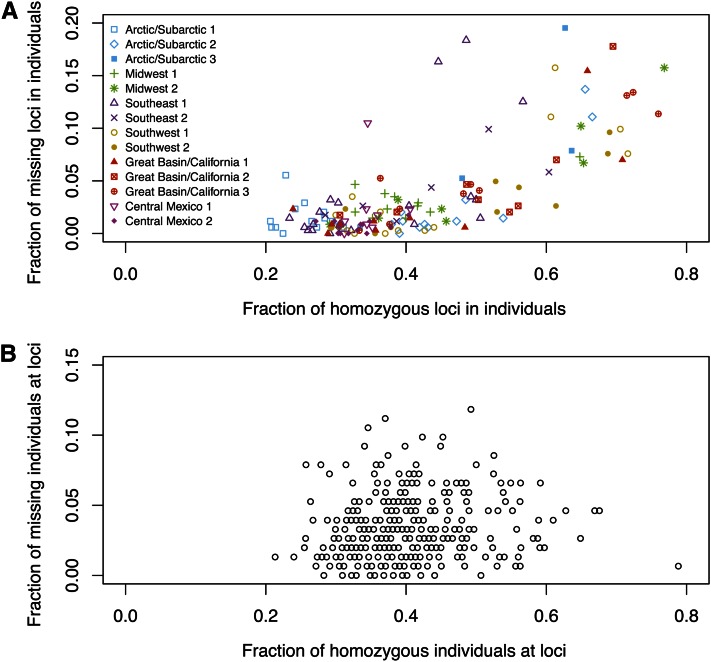
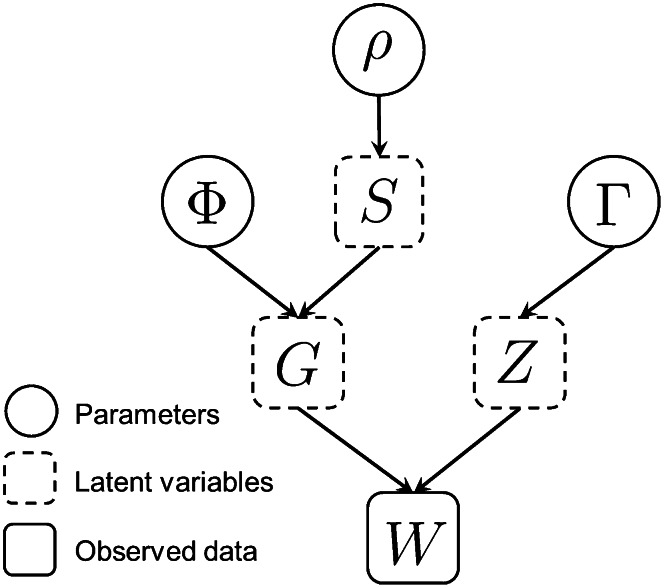
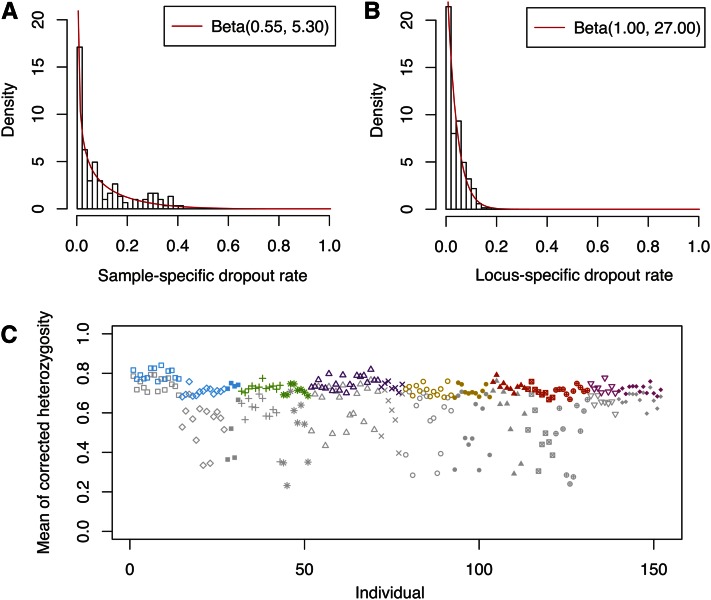
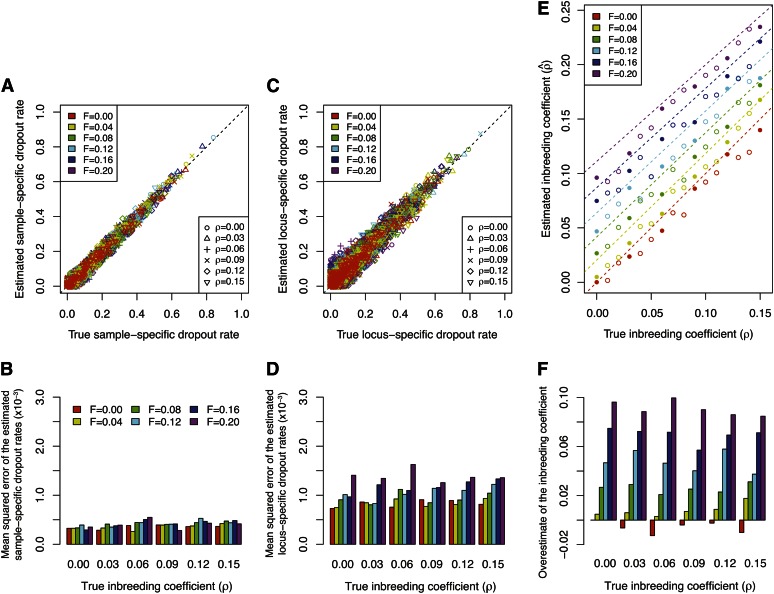
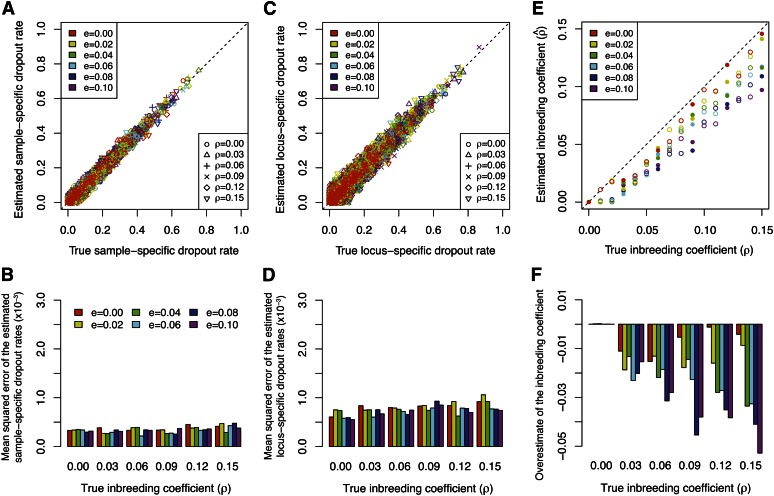
Allelic dropout is a commonly observed source of missing data in microsatellite genotypes, in which one or both allelic copies at a locus fail to be amplified by the polymerase chain reaction. Especially for samples with poor DNA quality, this problem causes a downward bias in estimates of observed heterozygosity and an upward bias in estimates of inbreeding, owing to mistaken classifications of heterozygotes as homozygotes when one of the two copies drops out. One general approach for avoiding allelic dropout involves repeated genotyping of homozygous loci to minimize the effects of experimental error. Existing computational alternatives often require replicate genotyping as well. These approaches, however, are costly and are suitable only when enough DNA is available for repeated genotyping. In this study, we propose a maximum-likelihood approach together with an expectation-maximization algorithm to jointly estimate allelic dropout rates and allele frequencies when only one set of nonreplicated genotypes is available. Our method considers estimates of allelic dropout caused by both sample-specific factors and locus-specific factors, and it allows for deviation from Hardy-Weinberg equilibrium owing to inbreeding. Using the estimated parameters, we correct the bias in the estimation of observed heterozygosity through the use of multiple imputations of alleles in cases where dropout might have occurred. With simulated data, we show that our method can (1) effectively reproduce patterns of missing data and heterozygosity observed in real data; (2) correctly estimate model parameters, including sample-specific dropout rates, locus-specific dropout rates, and the inbreeding coefficient; and (3) successfully correct the downward bias in estimating the observed heterozygosity. We find that our method is fairly robust to violations of model assumptions caused by population structure and by genotyping errors from sources other than allelic dropout. Because the data sets imputed under our model can be investigated in additional subsequent analyses, our method will be useful for preparing data for applications in diverse contexts in population genetics and molecular ecology.
Figures
References
-
- Bonin A., Bellemain E., Eidesen P. B., Pompanon F., Brochmann C., et al. , 2004. How to track and assess genotyping errors in population genetics studies. Mol. Ecol. 13: 3261–3273 - PubMed
-
- Broquet T., Petit E., 2004. Quantifying genotyping errors in noninvasive population genetics. Mol. Ecol. 13: 3601–3608 - PubMed
-
- Broquet T., Ménard N., Petit E., 2007. Noninvasive population genetics: a review of sample source, diet, fragment length and microsatellite motif effects on amplification success and genotyping error rates. Conserv. Genet. 8: 249–260
-
- Buchan J. C., Archie E. A., van Horn R. C., Moss C. J., Alberts S. C., 2005. Locus effects and sources of error in noninvasive genotyping. Mol. Ecol. Notes 5: 680–683
-
- Casella G., Berger R. L., 2001. Statistical Inference, Ed. 2 Duxbury, Pacific Grove, CA
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
