

Quantitative RNA-Seq analysis in non-model species: assessing transcriptome assemblies as a scaffold and the utility of evolutionary divergent genomic reference species
- PMID: 22853326
- PMCID: PMC3469347
- DOI: 10.1186/1471-2164-13-361
Quantitative RNA-Seq analysis in non-model species: assessing transcriptome assemblies as a scaffold and the utility of evolutionary divergent genomic reference species
Abstract
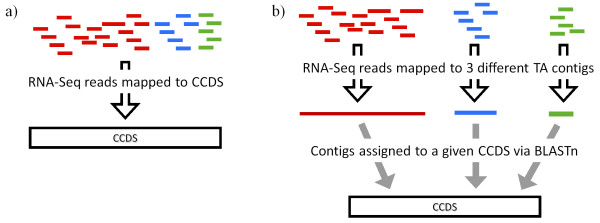
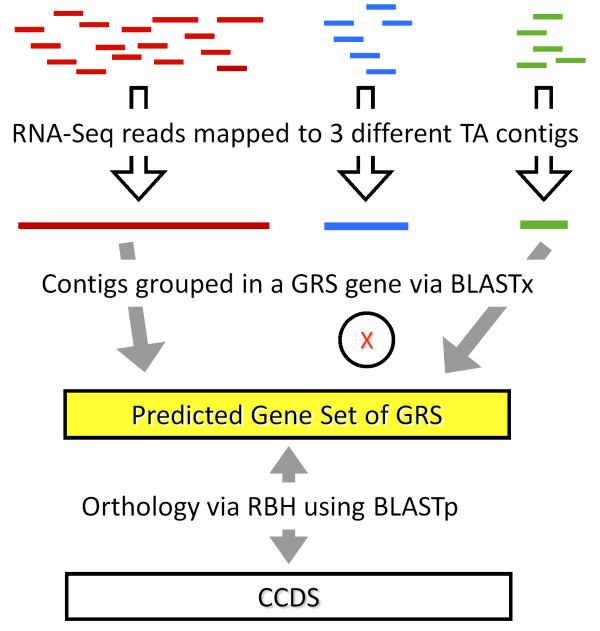
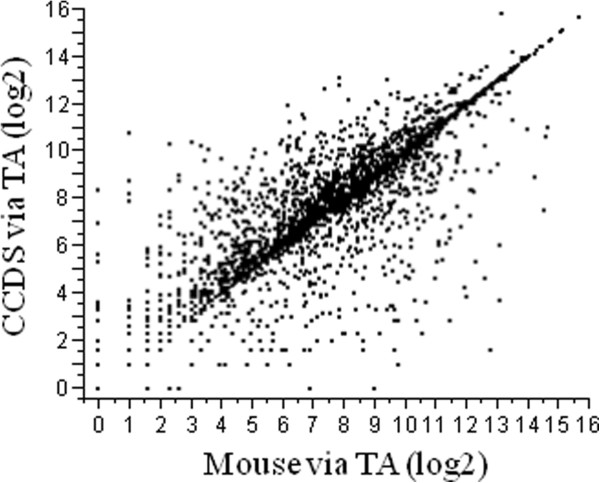
Background: How well does RNA-Seq data perform for quantitative whole gene expression analysis in the absence of a genome? This is one unanswered question facing the rapidly growing number of researchers studying non-model species. Using Homo sapiens data and resources, we compared the direct mapping of sequencing reads to predicted genes from the genome with mapping to de novo transcriptomes assembled from RNA-Seq data. Gene coverage and expression analysis was further investigated in the non-model context by using increasingly divergent genomic reference species to group assembled contigs by unique genes.
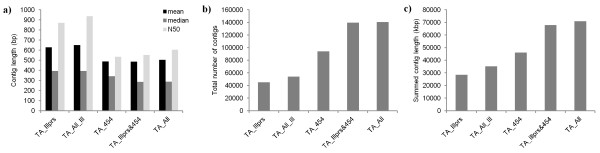
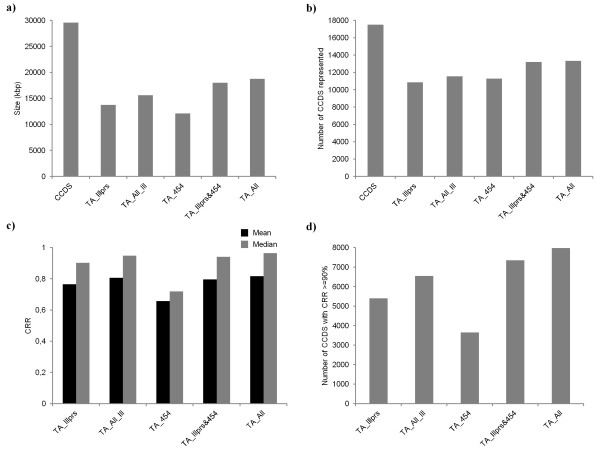
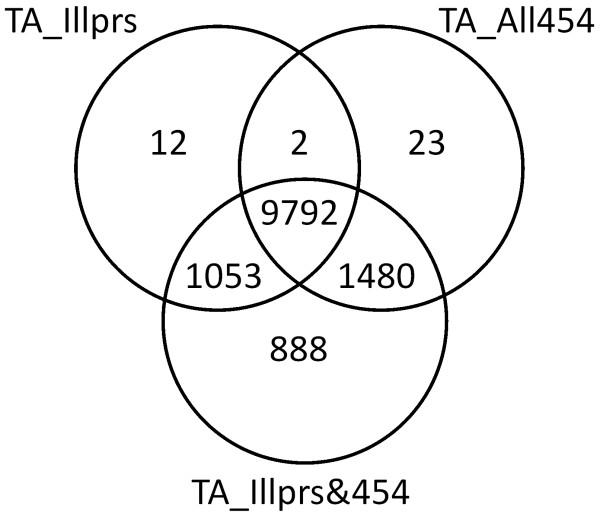
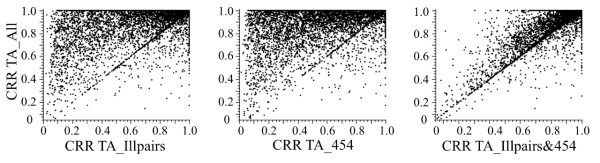
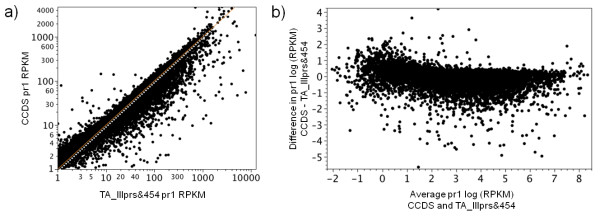
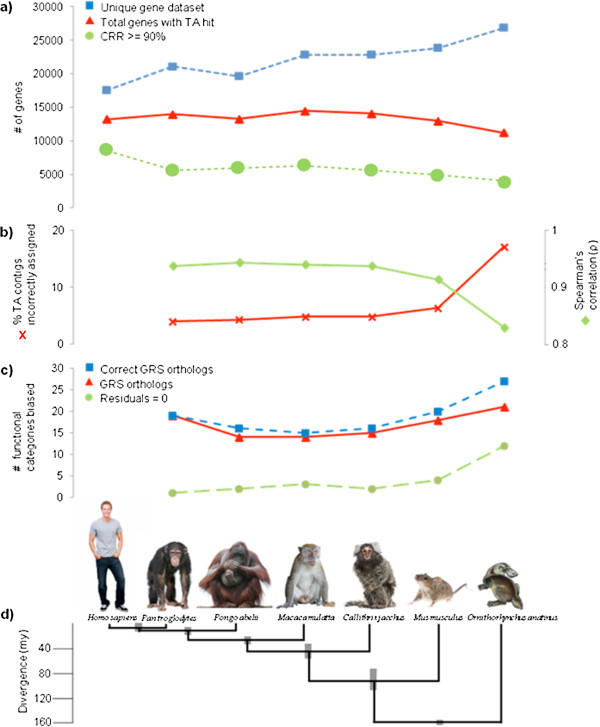
Results: Eight transcriptome sets, composed of varying amounts of Illumina and 454 data, were assembled and assessed. Hybrid 454/Illumina assemblies had the highest transcriptome and individual gene coverage. Quantitative whole gene expression levels were highly similar between using a de novo hybrid assembly and the predicted genes as a scaffold, although mapping to the de novo transcriptome assembly provided data on fewer genes. Using non-target species as reference scaffolds does result in some loss of sequence and expression data, and bias and error increase with evolutionary distance. However, within a 100 million year window these effect sizes are relatively small.
Conclusions: Predicted gene sets from sequenced genomes of related species can provide a powerful method for grouping RNA-Seq reads and annotating contigs. Gene expression results can be produced that are similar to results obtained using gene models derived from a high quality genome, though biased towards conserved genes. Our results demonstrate the power and limitations of conducting RNA-Seq in non-model species.
Figures
References
-
- Trapnell C, Williams B, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg S, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology. 2010;28:511–515. doi: 10.1038/nbt.1621. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
