

Insights from free-energy calculations: protein conformational equilibrium, driving forces, and ligand-binding modes
- PMID: 22853912
- PMCID: PMC3400776
- DOI: 10.1016/j.bpj.2012.05.046
Insights from free-energy calculations: protein conformational equilibrium, driving forces, and ligand-binding modes
Abstract
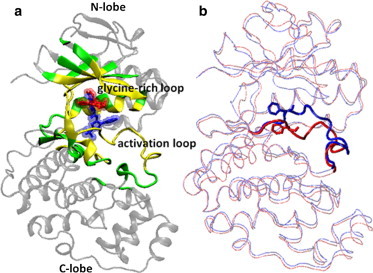
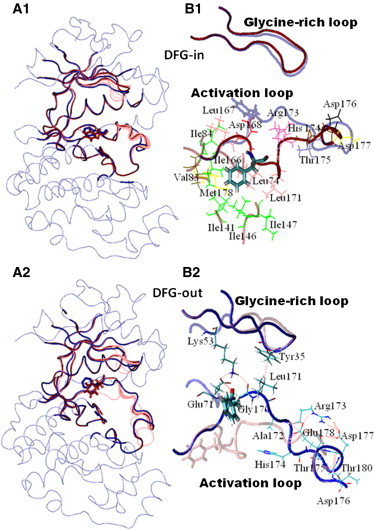
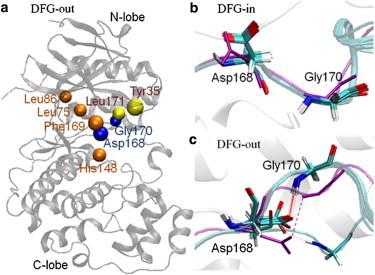
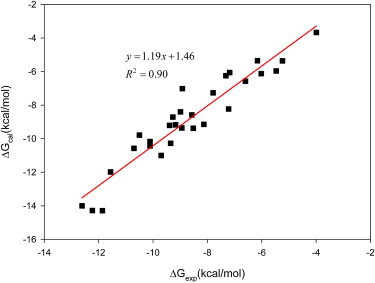
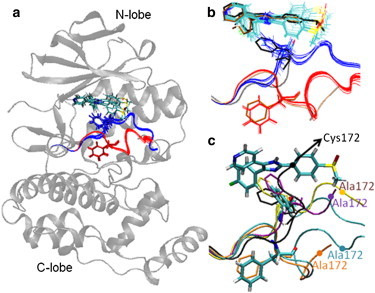
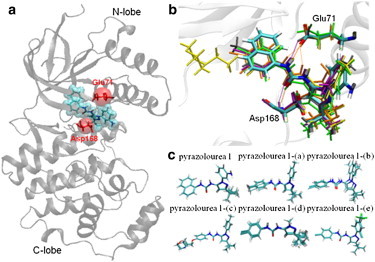
Accurate free-energy calculations provide mechanistic insights into molecular recognition and conformational equilibrium. In this work, we performed free-energy calculations to study the thermodynamic properties of different states of molecular systems in their equilibrium basin, and obtained accurate absolute binding free-energy calculations for protein-ligand binding using a newly developed M2 algorithm. We used a range of Asp-Phe-Gly (DFG)-in/out p38α mitogen-activated protein kinase inhibitors as our test cases. We also focused on the flexible DFG motif, which is closely connected to kinase activation and inhibitor binding. Our calculations explain the coexistence of DFG-in and DFG-out states of the loop and reveal different components (e.g., configurational entropy and enthalpy) that stabilize the apo p38α conformations. To study novel ligand-binding modes and the key driving forces behind them, we computed the absolute binding free energies of 30 p38α inhibitors, including analogs with unavailable experimental structures. The calculations revealed multiple stable, complex conformations and changes in p38α and inhibitor conformations, as well as balance in several energetic terms and configurational entropy loss. The results provide relevant physics that can aid in designing inhibitors and understanding protein conformational equilibrium. Our approach is fast for use with proteins that contain flexible regions for structure-based drug design.
Copyright © 2012 Biophysical Society. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Human p38α mitogen-activated protein kinase in the Asp168-Phe169-Gly170-in (DFG-in) state can bind allosteric inhibitor Doramapimod.J Biomol Struct Dyn. 2019 May;37(8):2049-2060. doi: 10.1080/07391102.2018.1475260. Epub 2018 Dec 5. J Biomol Struct Dyn. 2019. PMID: 29749295
-
Molecular dynamics simulation and free energy calculation studies of the binding mechanism of allosteric inhibitors with p38α MAP kinase.J Chem Inf Model. 2011 Dec 27;51(12):3235-46. doi: 10.1021/ci200159g. Epub 2011 Nov 18. J Chem Inf Model. 2011. PMID: 22097958
-
Fluorescence polarization binding assay to develop inhibitors of inactive p38alpha mitogen-activated protein kinase.Anal Biochem. 2010 Jun 1;401(1):125-33. doi: 10.1016/j.ab.2010.02.016. Epub 2010 Feb 20. Anal Biochem. 2010. PMID: 20175985
-
A Comprehensive Structural Overview of p38α MAPK in Complex with Type I Inhibitors.ChemMedChem. 2015 Jun;10(6):957-69. doi: 10.1002/cmdc.201500030. Epub 2015 Apr 9. ChemMedChem. 2015. PMID: 26012502 Review.
-
Pharmacophore design of p38α MAP kinase inhibitors with either 2,4,5-trisubstituted or 1,2,4,5-tetrasubstituted imidazole scaffold.Curr Med Chem. 2011;18(10):1526-39. doi: 10.2174/092986711795328409. Curr Med Chem. 2011. PMID: 21428890 Review.
Cited by
-
Bringing Clarity to the Prediction of Protein-Ligand Binding Free Energies via "Blurring".J Chem Theory Comput. 2014 Mar 11;10(3):1314-1325. doi: 10.1021/ct400995c. Epub 2014 Feb 7. J Chem Theory Comput. 2014. PMID: 24803861 Free PMC article.
-
Best practices for constructing, preparing, and evaluating protein-ligand binding affinity benchmarks [Article v0.1].Living J Comput Mol Sci. 2022;4(1):1497. doi: 10.33011/livecoms.4.1.1497. Epub 2022 Aug 30. Living J Comput Mol Sci. 2022. PMID: 36382113 Free PMC article.
-
Discovery of CDK8/CycC Ligands with a New Virtual Screening Tool.ChemMedChem. 2019 Jan 8;14(1):107-118. doi: 10.1002/cmdc.201800559. Epub 2018 Dec 10. ChemMedChem. 2019. PMID: 30403831 Free PMC article.
-
Uncovering water effects in protein-ligand recognition: importance in the second hydration shell and binding kinetics.Phys Chem Chem Phys. 2023 Jan 18;25(3):2098-2109. doi: 10.1039/d2cp04584b. Phys Chem Chem Phys. 2023. PMID: 36562309 Free PMC article.
-
Understanding ligand-receptor non-covalent binding kinetics using molecular modeling.Front Biosci (Landmark Ed). 2017 Jan 1;22(6):960-981. doi: 10.2741/4527. Front Biosci (Landmark Ed). 2017. PMID: 27814657 Free PMC article. Review.
References
-
- Huse M., Kuriyan J. The conformational plasticity of protein kinases. Cell. 2002;109:275–282. - PubMed
-
- Bert K., Gerhard M., Michael H., editors. Protein Kinases as Drug Targets. Wiley-VCH; Weinheim: 2011.
-
- Vogtherr M., Saxena K., Schwalbe H. NMR characterization of kinase p38 dynamics in free and ligand-bound forms. Angew. Chem. Int. Ed. Engl. 2006;45:993–997. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
