

Inhibition of apoptosis by BCL2 prevents leukemic transformation of a murine myelodysplastic syndrome
- PMID: 22855610
- PMCID: PMC3448259
- DOI: 10.1182/blood-2012-05-430736
Inhibition of apoptosis by BCL2 prevents leukemic transformation of a murine myelodysplastic syndrome
Abstract
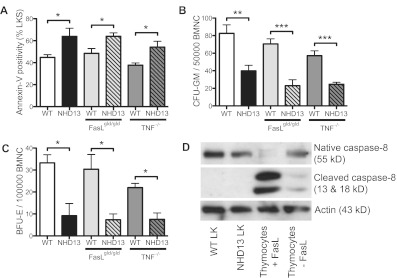
Programmed cell death or apoptosis is a prominent feature of low-risk myelodysplastic syndromes (MDS), although the underlying mechanism remains controversial. High-risk MDS have less apoptosis associated with increased expression of the prosurvival BCL2-related proteins. To address the mechanism and pathogenic role of apoptosis and BCL2 expression in MDS, we used a mouse model resembling human MDS, in which the fusion protein NUP98-HOXD13 (NHD13) of the chromosomal translocation t(2;11)(q31;p15) is expressed in hematopoietic cells. Hematopoietic stem and progenitor cells from 3-month-old mice had increased rates of apoptosis associated with increased cell cycling and DNA damage. Gene expression profiling of these MDS progenitors revealed a specific reduction in Bcl2. Restoration of Bcl2 expression by a BCL2 transgene blocked apoptosis of the MDS progenitors, which corrected the macrocytic anemia. Blocking apoptosis also restored cell-cycle quiescence and reduced DNA damage in the MDS progenitors. We expected that preventing apoptosis would accelerate malignant transformation to acute myeloid leukemia (AML). However, contrary to expectations, preventing apoptosis of premalignant cells abrogated transformation to AML. In contrast to the current dogma that overcoming apoptosis is an important step toward cancer, this work demonstrates that gaining a survival advantage of premalignant cells may delay or prevent leukemic progression.
Figures





References
-
- Tefferi A, Vardiman JW. Myelodysplastic syndromes. N Engl J Med. 2009;361(19):1872–1885. - PubMed
-
- Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9(1):47–59. - PubMed
-
- Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319(5868):1352–1355. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
