

Cell-type specificity of ChIP-predicted transcription factor binding sites
- PMID: 22863112
- PMCID: PMC3574057
- DOI: 10.1186/1471-2164-13-372
Cell-type specificity of ChIP-predicted transcription factor binding sites
Abstract
Background: Context-dependent transcription factor (TF) binding is one reason for differences in gene expression patterns between different cellular states. Chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-seq) identifies genome-wide TF binding sites for one particular context-the cells used in the experiment. But can such ChIP-seq data predict TF binding in other cellular contexts and is it possible to distinguish context-dependent from ubiquitous TF binding?
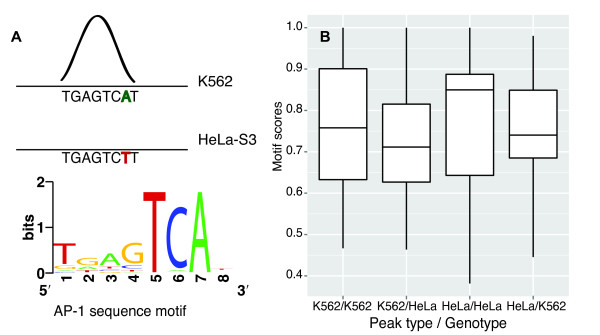
Results: We compared ChIP-seq data on TF binding for multiple TFs in two different cell types and found that on average only a third of ChIP-seq peak regions are common to both cell types. Expectedly, common peaks occur more frequently in certain genomic contexts, such as CpG-rich promoters, whereas chromatin differences characterize cell-type specific TF binding. We also find, however, that genotype differences between the cell types can explain differences in binding. Moreover, ChIP-seq signal intensity and peak clustering are the strongest predictors of common peaks. Compared with strong peaks located in regions containing peaks for multiple transcription factors, weak and isolated peaks are less common between the cell types and are less associated with data that indicate regulatory activity.
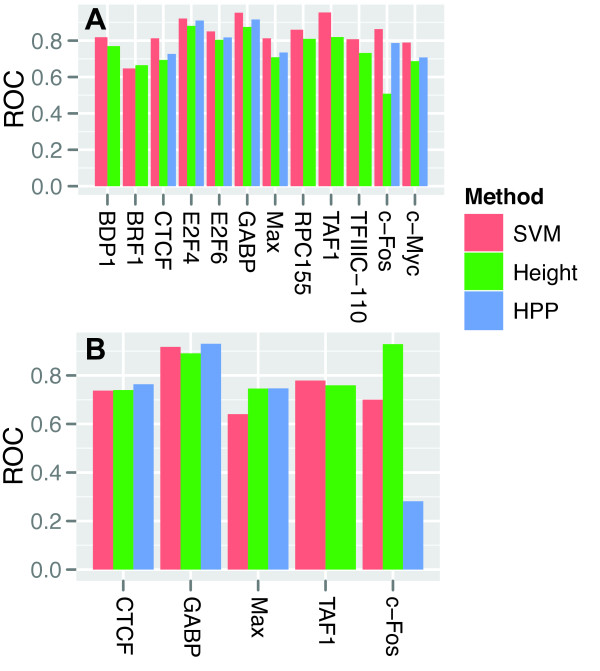
Conclusions: Together, the results suggest that experimental noise is prevalent among weak peaks, whereas strong and clustered peaks represent high-confidence binding events that often occur in other cellular contexts. Nevertheless, 30-40% of the strongest and most clustered peaks show context-dependent regulation. We show that by combining signal intensity with additional data-ranging from context independent information such as binding site conservation and position weight matrix scores to context dependent chromatin structure-we can predict whether a ChIP-seq peak is likely to be present in other cellular contexts.
Figures






References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
