

A single recurrent mutation in the 5'-UTR of IFITM5 causes osteogenesis imperfecta type V
- PMID: 22863190
- PMCID: PMC3415533
- DOI: 10.1016/j.ajhg.2012.06.005
A single recurrent mutation in the 5'-UTR of IFITM5 causes osteogenesis imperfecta type V
Abstract
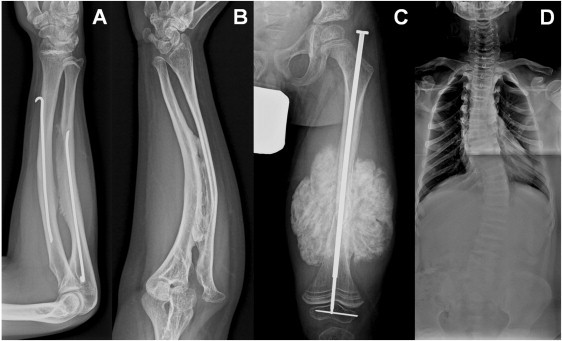
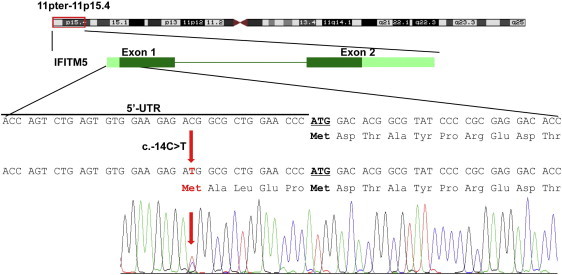
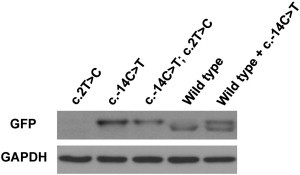
Osteogenesis imperfecta (OI) is a heterogenous group of genetic disorders of bone fragility. OI type V is an autosomal-dominant disease characterized by calcification of the forearm interosseous membrane, radial head dislocation, a subphyseal metaphyseal radiodense line, and hyperplastic callus formation; the causative mutation involved in this disease has not been discovered yet. Using linkage analysis in a four-generation family and whole-exome sequencing, we identified a heterozygous mutation of c.-14C>T in the 5'-untranslated region of a gene encoding interferon-induced transmembrane protein 5 (IFITM5). It completely cosegregated with the disease in three families and occurred de novo in five simplex individuals. Transfection of wild-type and mutant IFITM5 constructs revealed that the mutation added five amino acids (Met-Ala-Leu-Glu-Pro) to the N terminus of IFITM5. Given that IFITM5 expression and protein localization is restricted to the skeletal tissue and IFITM5 involvement in bone formation, we conclude that this recurrent mutation would have a specific effect on IFITM5 function and thus cause OI type V.
Copyright © 2012 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Rauch F., Glorieux F.H. Osteogenesis imperfecta. Lancet. 2004;363:1377–1385. - PubMed
-
- Morello R., Bertin T.K., Chen Y., Hicks J., Tonachini L., Monticone M., Castagnola P., Rauch F., Glorieux F.H., Vranka J. CRTAP is required for prolyl 3- hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell. 2006;127:291–304. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
