

A mutation in the 5'-UTR of IFITM5 creates an in-frame start codon and causes autosomal-dominant osteogenesis imperfecta type V with hyperplastic callus
- PMID: 22863195
- PMCID: PMC3415541
- DOI: 10.1016/j.ajhg.2012.06.011
A mutation in the 5'-UTR of IFITM5 creates an in-frame start codon and causes autosomal-dominant osteogenesis imperfecta type V with hyperplastic callus
Abstract
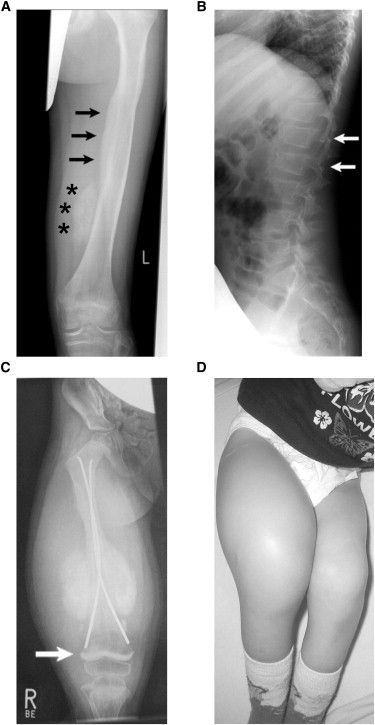
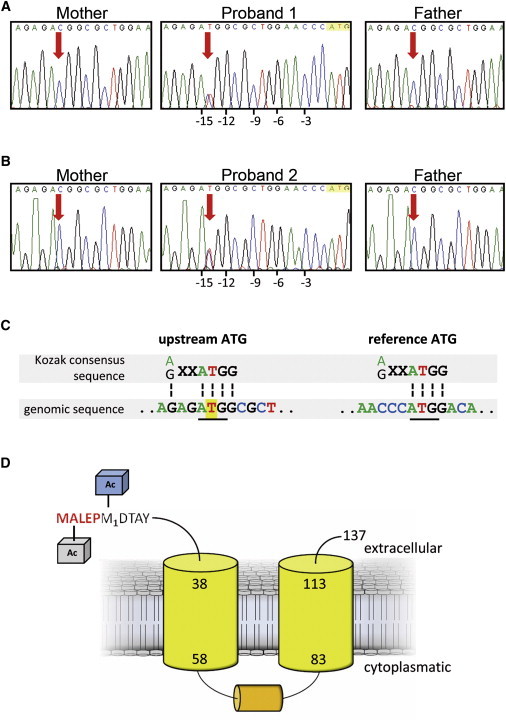
Osteogenesis imperfecta (OI) is a clinically and genetically heterogeneous disorder associated with bone fragility and susceptibility to fractures after minimal trauma. OI type V has an autosomal-dominant pattern of inheritance and is not caused by mutations in the type I collagen genes COL1A1 and COL1A2. The most remarkable and pathognomonic feature, observed in ~65% of affected individuals, is a predisposition to develop hyperplastic callus after fractures or surgical interventions. To identify the molecular cause of OI type V, we performed whole-exome sequencing in a female with OI type V and her unaffected parents and searched for de novo mutations. We found a heterozygous de novo mutation in the 5'-untranslated region of IFITM5 (the gene encoding Interferon induced transmembrane protein 5), 14 bp upstream of the annotated translation initiation codon (c.-14C>T). Subsequently, we identified an identical heterozygous de novo mutation in a second individual with OI type V by Sanger sequencing, thereby confirming that this is the causal mutation for the phenotype. IFITM5 is a protein that is highly enriched in osteoblasts and has a putative function in bone formation and osteoblast maturation. The mutation c.-14C>T introduces an upstream start codon that is in frame with the reference open-reading frame of IFITM5 and is embedded into a stronger Kozak consensus sequence for translation initiation than the annotated start codon. In vitro, eukaryotic cells were able to recognize this start codon, and they used it instead of the reference translation initiation signal. This suggests that five amino acids (Met-Ala-Leu-Glu-Pro) are added to the N terminus and alter IFITM5 function in individuals with the mutation.
Copyright © 2012 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures

References
-
- Chu M.L., Williams C.J., Pepe G., Hirsch J.L., Prockop D.J., Ramirez F. Internal deletion in a collagen gene in a perinatal lethal form of osteogenesis imperfecta. Nature. 1983;304:78–80. - PubMed
-
- Williams C.J., Prockop D.J. Synthesis and processing of a type I procollagen containing shortened pro-alpha 1(I) chains by fibroblasts from a patient with osteogenesis imperfecta. J. Biol. Chem. 1983;258:5915–5921. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
- RC2 HL102926/HL/NHLBI NIH HHS/United States
- HL-102924/HL/NHLBI NIH HHS/United States
- RC2 HL102924/HL/NHLBI NIH HHS/United States
- HL-102926/HL/NHLBI NIH HHS/United States
- HL-102925/HL/NHLBI NIH HHS/United States
- HL-102923/HL/NHLBI NIH HHS/United States
- RC2 HL102923/HL/NHLBI NIH HHS/United States
- UC2 HL102926/HL/NHLBI NIH HHS/United States
- UC2 HL103010/HL/NHLBI NIH HHS/United States
- HL-103010/HL/NHLBI NIH HHS/United States
- UC2 HL102923/HL/NHLBI NIH HHS/United States
- UC2 HL102924/HL/NHLBI NIH HHS/United States
- RC2 HL103010/HL/NHLBI NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- RC2 HL102925/HL/NHLBI NIH HHS/United States
- UC2 HL102925/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
