

Transforming clinical microbiology with bacterial genome sequencing
- PMID: 22868263
- PMCID: PMC5049685
- DOI: 10.1038/nrg3226
Transforming clinical microbiology with bacterial genome sequencing
Abstract
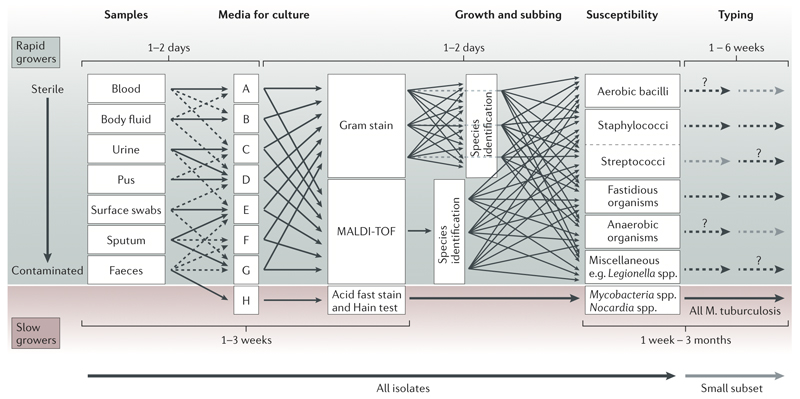
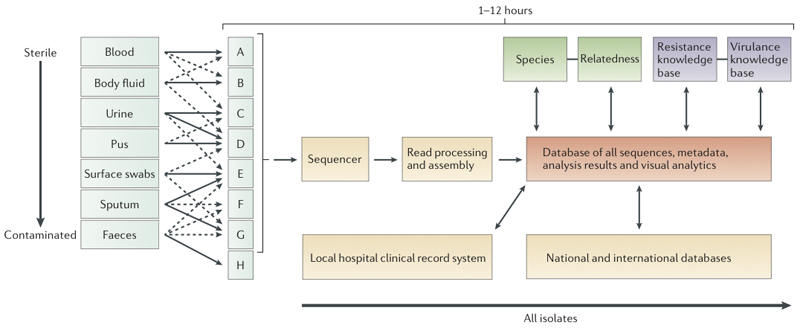
Whole-genome sequencing of bacteria has recently emerged as a cost-effective and convenient approach for addressing many microbiological questions. Here, we review the current status of clinical microbiology and how it has already begun to be transformed by using next-generation sequencing. We focus on three essential tasks: identifying the species of an isolate, testing its properties, such as resistance to antibiotics and virulence, and monitoring the emergence and spread of bacterial pathogens. We predict that the application of next-generation sequencing will soon be sufficiently fast, accurate and cheap to be used in routine clinical microbiology practice, where it could replace many complex current techniques with a single, more efficient workflow.
Figures
References
-
- Burlage RS. Principles of public health microbiology. Jones & Bartlett Learning; Sudbury, MA: 2012.
-
- Mandell GL, Bennett JE, Dolin R. Mandell, Douglas, and Bennett's principles and practice of infectious diseases. Churchill Livingstone/Elsevier; Philadelphia, PA: 2010.
-
- Murray PR, Rosenthal KS, Pfaller MA. Medical microbiology. Mosby/Elsevier; Philadelphia: 2009.
Highlighted References
-
-
Konstantinidis, K.T. & Tiedje, J.M. Genomic insights that advance the species definition for prokaryotes. Proc Natl Acad Sci U S A 102, 2567-72 (2005).
- [First description of a computation criteria to define bacterial species based on whole-genome sequencing.]
-
-
-
Jolley, K.A. & Maiden, M.C. BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 11, 595 (2010).
- [A database system for whole genomes that provides a smooth transition for users from working with MLST to working with genomes.]
-
-
-
Bille, E. et al. A chromosomally integrated bacteriophage in invasive meningococci. J Exp Med 201, 1905-13 (2005).
- [First example of an association mapping study to determine virulence factors in Neisseria meningitidis.]
-
-
-
Young, B.C. et al. Evolutionary dynamics of Staphylococcus aureus during progression from carriage to disease. Proc Natl Acad Sci U S A 109, 4550-5 (2012).
- [A detailed investigation of Staphylococcus aureus within-host genomic diversification in time revealing a probable evolution towards increased virulence.]
-
-
-
Rasko, D.A. et al. Origins of the E. coli strain causing an outbreak of hemolytic-uremic syndrome in Germany. N Engl J Med 365, 709-17 (2011).
- [Epidemiological investigation based on whole-genome sequencing for the 2011 German outbreak of Escherichia coli.]
-
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
