

Loss of ATM kinase activity leads to embryonic lethality in mice
- PMID: 22869595
- PMCID: PMC3413361
- DOI: 10.1083/jcb.201204035
Loss of ATM kinase activity leads to embryonic lethality in mice
Abstract
Ataxia telangiectasia (A-T) mutated (ATM) is a key deoxyribonucleic acid (DNA) damage signaling kinase that regulates DNA repair, cell cycle checkpoints, and apoptosis. The majority of patients with A-T, a cancer-prone neurodegenerative disease, present with null mutations in Atm. To determine whether the functions of ATM are mediated solely by its kinase activity, we generated two mouse models containing single, catalytically inactivating point mutations in Atm. In this paper, we show that, in contrast to Atm-null mice, both D2899A and Q2740P mutations cause early embryonic lethality in mice, without displaying dominant-negative interfering activity. Using conditional deletion, we find that the D2899A mutation in adult mice behaves largely similar to Atm-null cells but shows greater deficiency in homologous recombination (HR) as measured by hypersensitivity to poly (adenosine diphosphate-ribose) polymerase inhibition and increased genomic instability. These results may explain why missense mutations with no detectable kinase activity are rarely found in patients with classical A-T. We propose that ATM kinase-inactive missense mutations, unless otherwise compensated for, interfere with HR during embryogenesis.
Figures
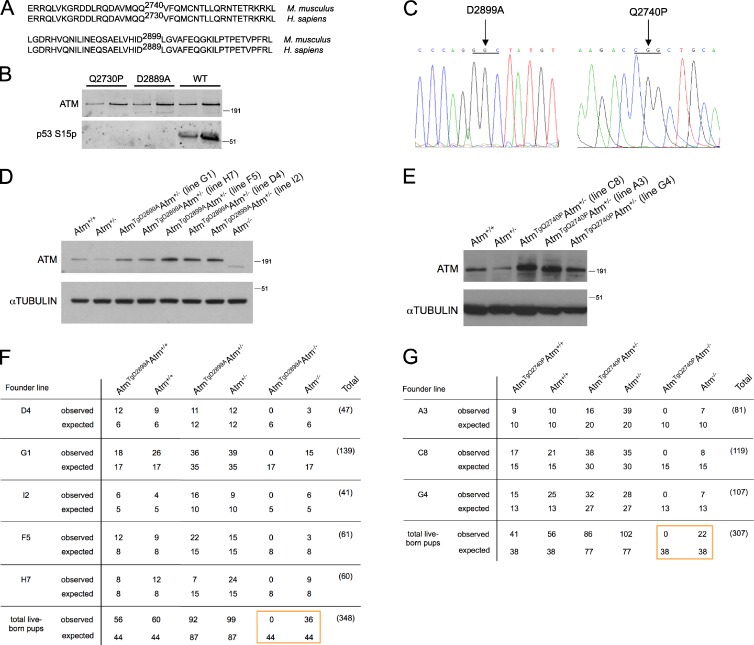
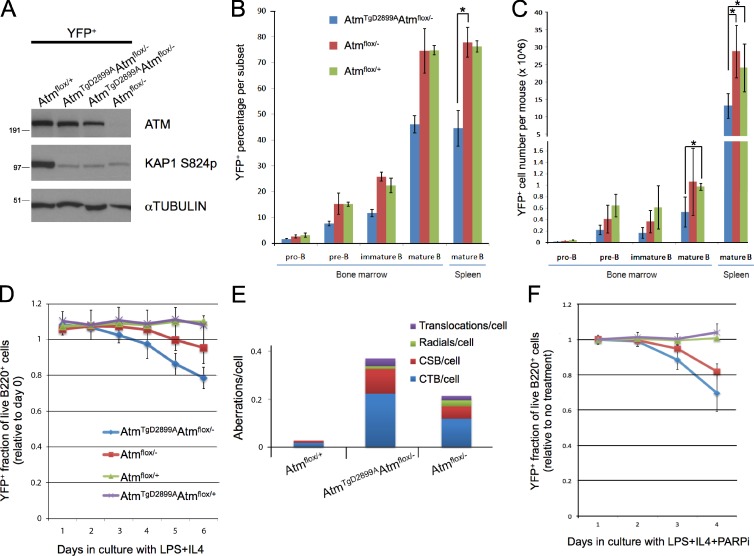
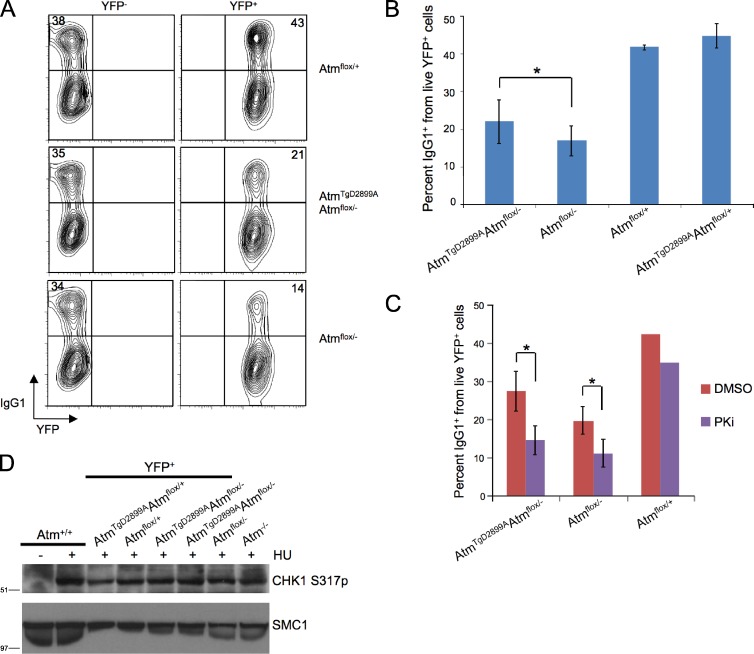
Comment in
-
The ATM protein: the importance of being active.J Cell Biol. 2012 Aug 6;198(3):273-5. doi: 10.1083/jcb.201207063. J Cell Biol. 2012. PMID: 22869592 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
