

BRCA1 and HSP90 cooperate in homologous and non-homologous DNA double-strand-break repair and G2/M checkpoint activation
- PMID: 22869732
- PMCID: PMC3427093
- DOI: 10.1073/pnas.1203326109
BRCA1 and HSP90 cooperate in homologous and non-homologous DNA double-strand-break repair and G2/M checkpoint activation
Abstract
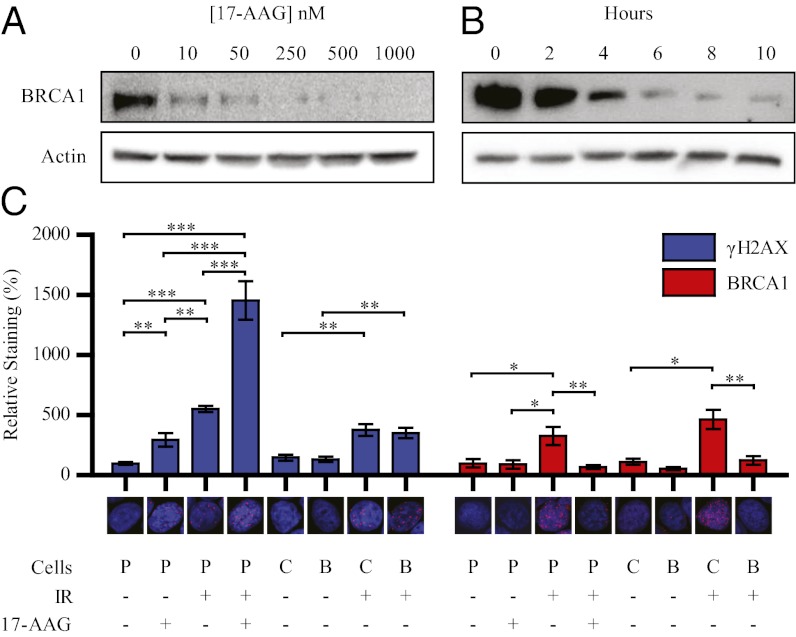
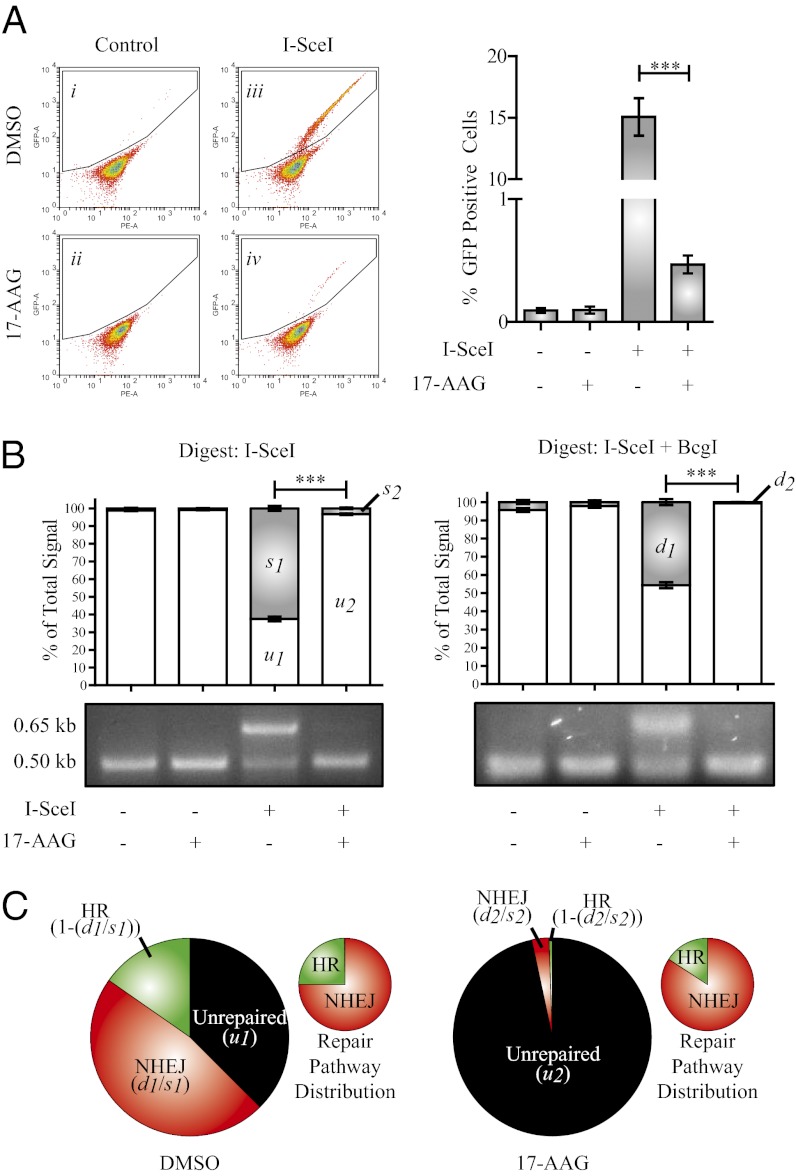
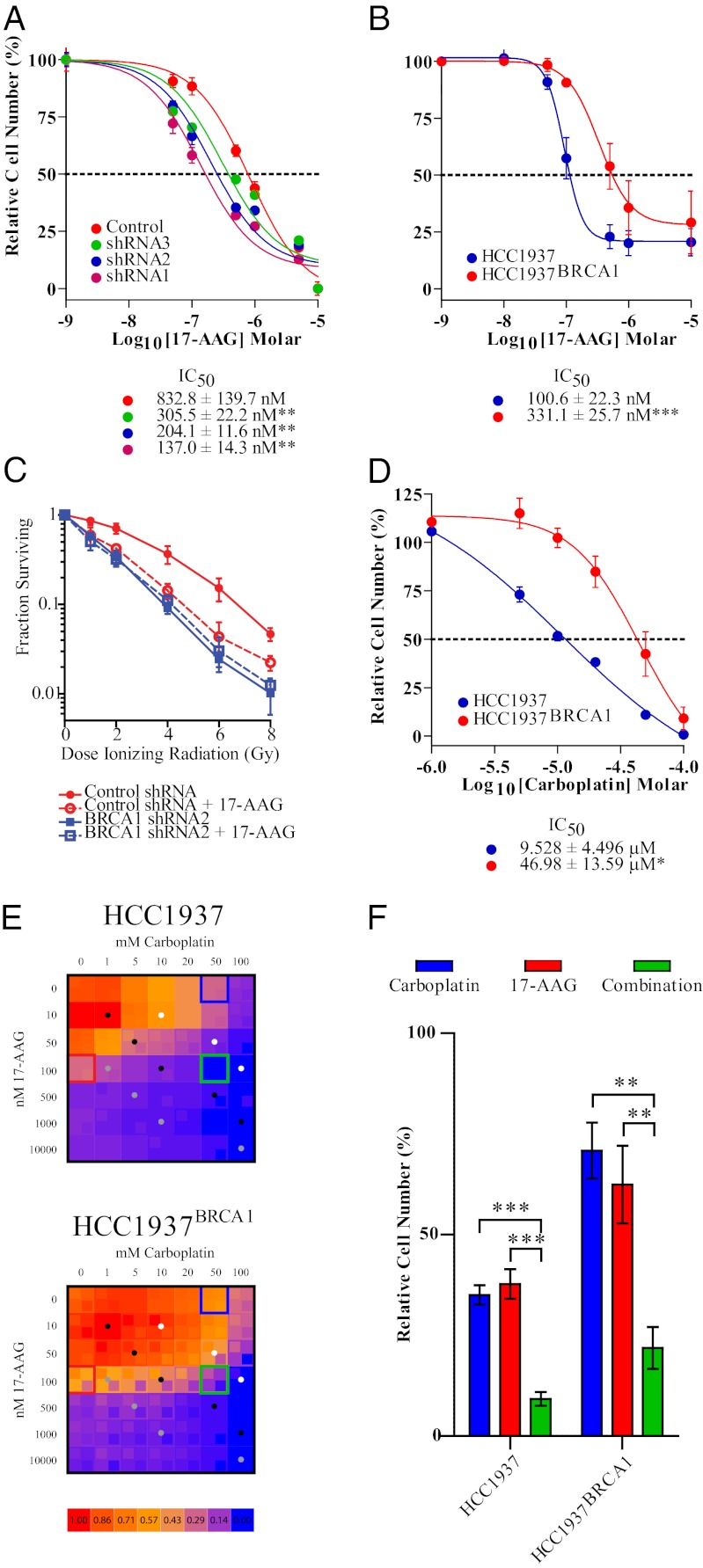
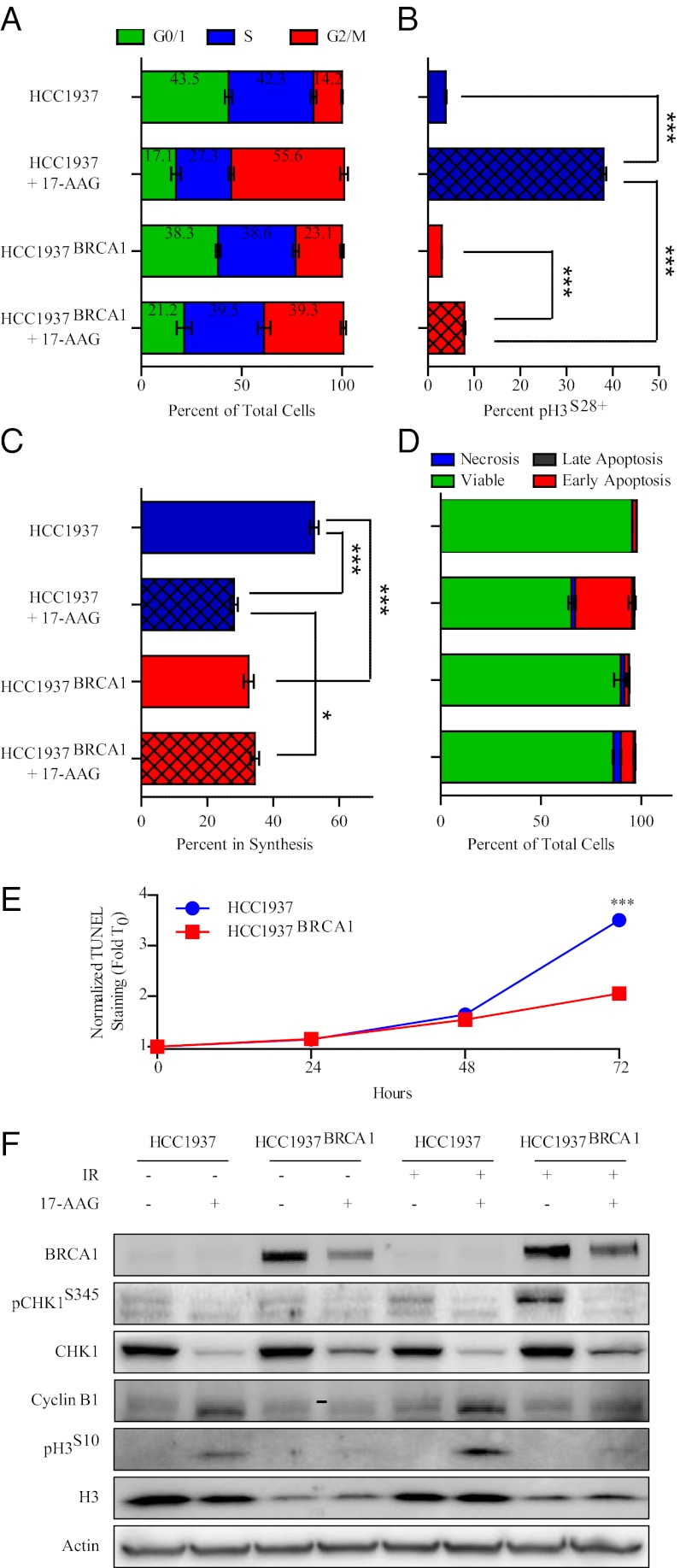
Expression of functional breast cancer susceptibility gene 1 (BRCA1) in human breast and ovarian cancers is associated with resistance to platinum-based chemotherapeutics and poly(ADP ribose) polymerase (PARP) inhibitors. BRCA1 is a nuclear tumor suppressor that is critical for resolving double-strand DNA breaks (DSBs) and interstrand crosslinks (ICLs) by homologous recombination (HR). In vitro, animal and human clinical data have demonstrated that BRCA1-deficient cancers are highly sensitive to ICL-inducing chemotherapeutic agents, are amenable to synthetic lethal approaches that exploit defects in DSB/ICL repair, and may be associated with improved survival. Conversely, high or restored expression of BRCA1 in breast and ovarian cancer is associated with therapeutic resistance and poor prognosis. There has been much interest in identifying agents that interfere with BRCA1-dependent DSB/ICL repair to restore or enhance sensitivity to cancer therapeutics. We demonstrate that the heat-shock protein 90 (HSP90) inhibitor 17-allylamino-17-demethoxygeldanamycin [17-AAG (Tanespimycin)], currently in Phase II/III clinical evaluation for several cancers, induces BRCA1 ubiquitination and proteasomal degradation, resulting in compromised repair of ionizing radiation- and platinum-induced DNA damage. We show that loss of HSP90 function abolishes BRCA1-dependent DSB repair and that BRCA1-deficient cells are hypersensitive to 17-AAG due to impaired Gap 2/Mitosis (G2/M) checkpoint activation and resultant mitotic catastrophe. In summary, we document an upstream HSP90-dependent regulatory point in the Fanconi anemia/BRCA DSB/ICL repair pathway, illuminate the role of BRCA1 in regulating damage-associated checkpoint and repair responses to HSP90 inhibitors, and identify BRCA1 as a clinically relevant target for enhancing sensitivity in refractory and/or resistant malignancies.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Ford D, Easton DF, Bishop DT, Narod SA, Goldgar DE. Breast Cancer Linkage Consortium Risks of cancer in BRCA1-mutation carriers. Lancet. 1994;343:692–695. - PubMed
-
- King MC, Marks JH, Mandell JB. New York Breast Cancer Study Group Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science. 2003;302:643–646. - PubMed
-
- Thompson ME, Jensen RA, Obermiller PS, Page DL, Holt JT. Decreased expression of BRCA1 accelerates growth and is often present during sporadic breast cancer progression. Nat Genet. 1995;9:444–450. - PubMed
-
- Hall JM, et al. Linkage of early-onset familial breast cancer to chromosome 17q21. Science. 1990;250:1684–1689. - PubMed
-
- Friedman LS, et al. Confirmation of BRCA1 by analysis of germline mutations linked to breast and ovarian cancer in ten families. Nat Genet. 1994;8:399–404. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
