

Computational design of catalytic dyads and oxyanion holes for ester hydrolysis
- PMID: 22871159
- PMCID: PMC4104585
- DOI: 10.1021/ja3037367
Computational design of catalytic dyads and oxyanion holes for ester hydrolysis
Abstract
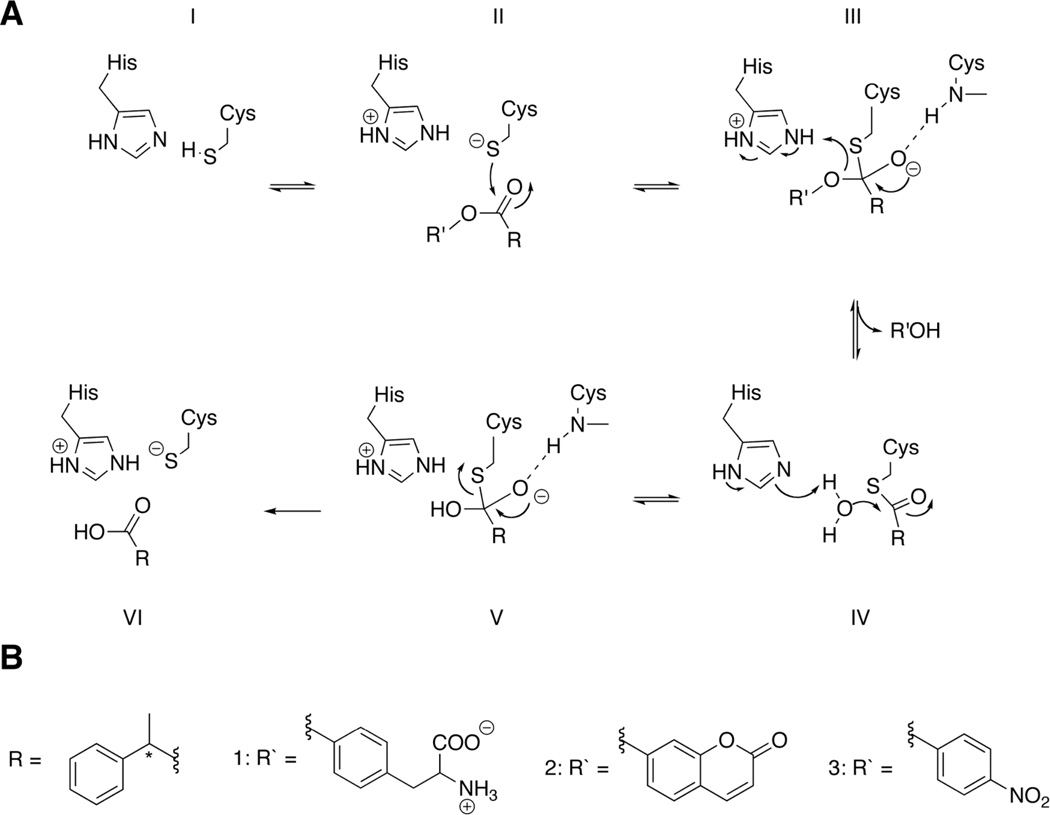
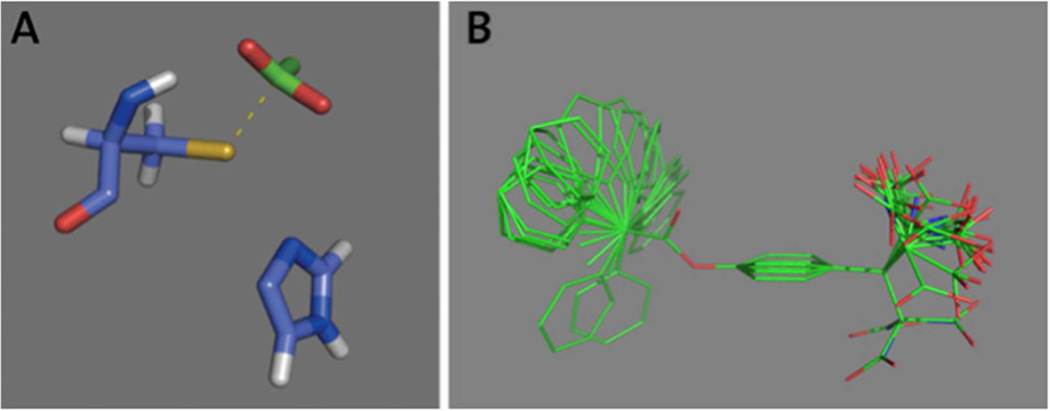
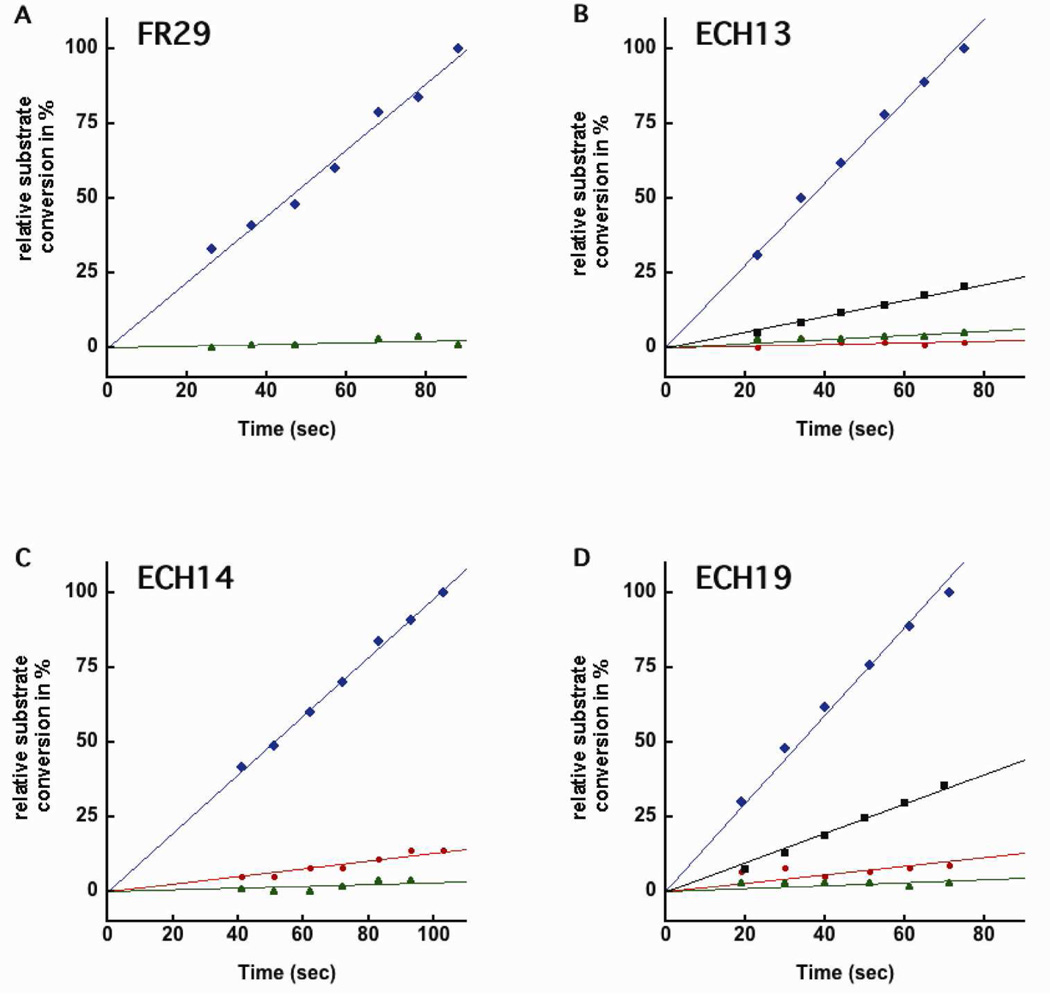
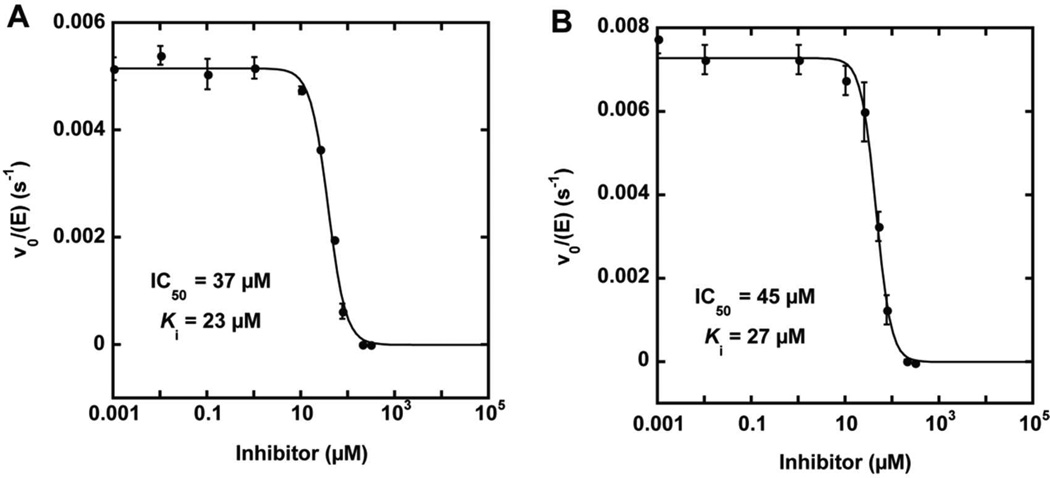
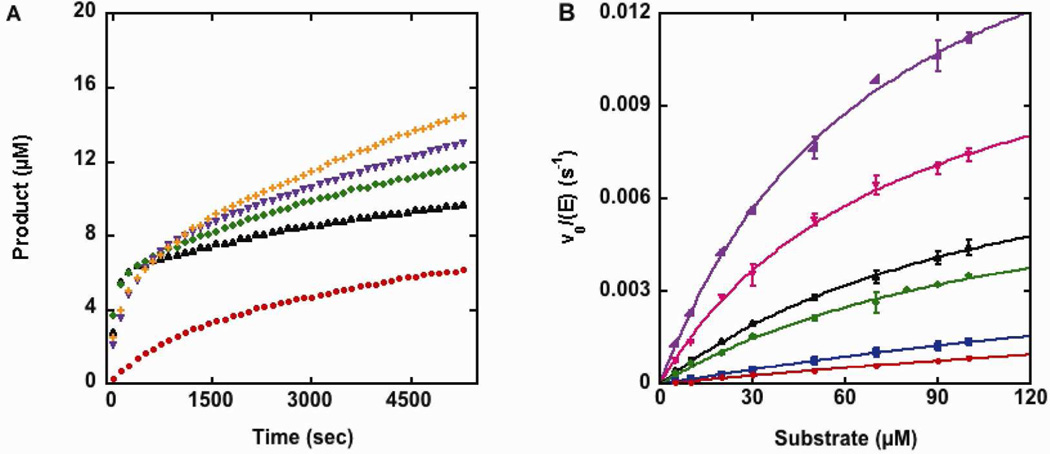
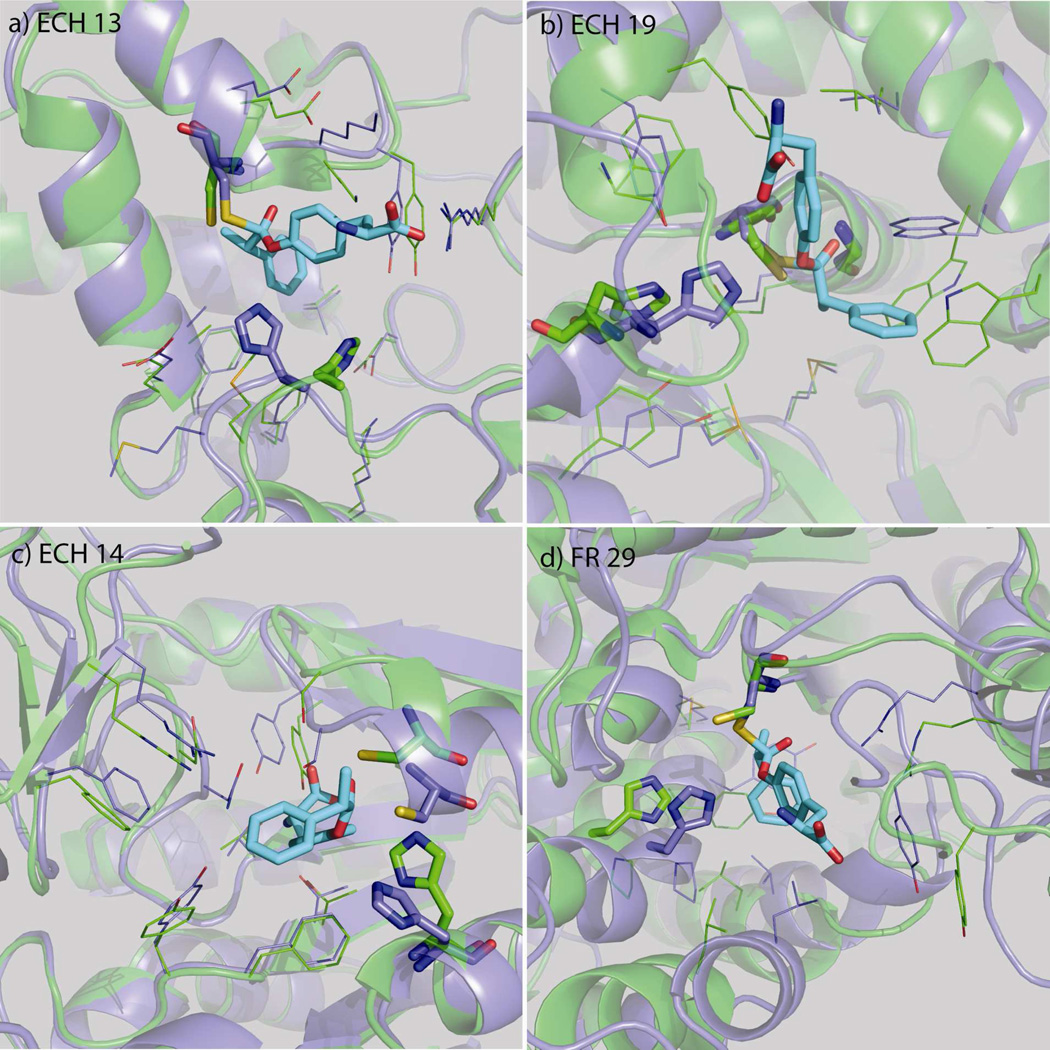
Nucleophilic catalysis is a general strategy for accelerating ester and amide hydrolysis. In natural active sites, nucleophilic elements such as catalytic dyads and triads are usually paired with oxyanion holes for substrate activation, but it is difficult to parse out the independent contributions of these elements or to understand how they emerged in the course of evolution. Here we explore the minimal requirements for esterase activity by computationally designing artificial catalysts using catalytic dyads and oxyanion holes. We found much higher success rates using designed oxyanion holes formed by backbone NH groups rather than by side chains or bridging water molecules and obtained four active designs in different scaffolds by combining this motif with a Cys-His dyad. Following active site optimization, the most active of the variants exhibited a catalytic efficiency (k(cat)/K(M)) of 400 M(-1) s(-1) for the cleavage of a p-nitrophenyl ester. Kinetic experiments indicate that the active site cysteines are rapidly acylated as programmed by design, but the subsequent slow hydrolysis of the acyl-enzyme intermediate limits overall catalytic efficiency. Moreover, the Cys-His dyads are not properly formed in crystal structures of the designed enzymes. These results highlight the challenges that computational design must overcome to achieve high levels of activity.
Figures
Similar articles
-
Investigation of a general base mechanism for ester hydrolysis in C-C hydrolase enzymes of the alpha/beta-hydrolase superfamily: a novel mechanism for the serine catalytic triad.Org Biomol Chem. 2007 Feb 7;5(3):507-13. doi: 10.1039/b615605c. Epub 2006 Dec 19. Org Biomol Chem. 2007. PMID: 17252134
-
The thiolase reaction mechanism: the importance of Asn316 and His348 for stabilizing the enolate intermediate of the Claisen condensation.Biochemistry. 2009 Nov 24;48(46):11011-25. doi: 10.1021/bi901069h. Biochemistry. 2009. PMID: 19842716
-
Facilitating the Evolution of Esterase Activity from a Promiscuous Enzyme (Mhg) with Catalytic Functions of Amide Hydrolysis and Carboxylic Acid Perhydrolysis by Engineering the Substrate Entrance Tunnel.Appl Environ Microbiol. 2016 Oct 27;82(22):6748-6756. doi: 10.1128/AEM.01817-16. Print 2016 Nov 15. Appl Environ Microbiol. 2016. PMID: 27613682 Free PMC article.
-
Distinction between esterases and lipases: comparative biochemical properties of sequence-related carboxylesterases.Protein Pept Lett. 2009;16(10):1149-61. doi: 10.2174/092986609789071333. Protein Pept Lett. 2009. PMID: 19508178 Review.
-
GDSL family of serine esterases/lipases.Prog Lipid Res. 2004 Nov;43(6):534-52. doi: 10.1016/j.plipres.2004.09.002. Prog Lipid Res. 2004. PMID: 15522763 Review.
Cited by
-
Systematic optimization model and algorithm for binding sequence selection in computational enzyme design.Protein Sci. 2013 Jul;22(7):929-41. doi: 10.1002/pro.2275. Epub 2013 Jun 6. Protein Sci. 2013. PMID: 23649589 Free PMC article.
-
Conformational dynamics and enzyme evolution.J R Soc Interface. 2018 Jul;15(144):20180330. doi: 10.1098/rsif.2018.0330. J R Soc Interface. 2018. PMID: 30021929 Free PMC article. Review.
-
Minimalistic Artificial Catalysts with Esterase-Like Activity from Multivalent Nanofibers Formed by the Self-Assembly of Dipeptides.ACS Omega. 2022 Dec 30;8(2):2491-2500. doi: 10.1021/acsomega.2c06972. eCollection 2023 Jan 17. ACS Omega. 2022. PMID: 36687071 Free PMC article.
-
TMEM164 is an acyltransferase that forms ferroptotic C20:4 ether phospholipids.Nat Chem Biol. 2023 Mar;19(3):378-388. doi: 10.1038/s41589-022-01253-7. Epub 2023 Feb 13. Nat Chem Biol. 2023. PMID: 36782012 Free PMC article.
-
An enumerative algorithm for de novo design of proteins with diverse pocket structures.Proc Natl Acad Sci U S A. 2020 Sep 8;117(36):22135-22145. doi: 10.1073/pnas.2005412117. Epub 2020 Aug 24. Proc Natl Acad Sci U S A. 2020. PMID: 32839327 Free PMC article.
References
-
- Clark JD, Schievella AR, Nalefski EA, Lin LL. J. Lipid Mediat. Cell. Signal. 1995;12:83–117. - PubMed
-
- Mignatti P, Rifkin DB. Enzyme Protein. 1996;49:117–137. - PubMed
-
- DeClerck YA, Imren S, Montgomery AM, Mueller BM, Reisfeld RA, Laug WE. Adv. Exp. Med. Biol. 1997;425:89–97. - PubMed
-
- MacDonald T, DeClerck Y, Laug W. Thromb. Haemostasis. 1997:S1541–S1541.
-
- Gorrell MD. Clin. Sci. (Lond) 2005;108:277–292. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
