

A probabilistic framework for peptide and protein quantification from data-dependent and data-independent LC-MS proteomics experiments
- PMID: 22871168
- PMCID: PMC3721451
- DOI: 10.1089/omi.2012.0019
A probabilistic framework for peptide and protein quantification from data-dependent and data-independent LC-MS proteomics experiments
Abstract
A probability-based quantification framework is presented for the calculation of relative peptide and protein abundance in label-free and label-dependent LC-MS proteomics data. The results are accompanied by credible intervals and regulation probabilities. The algorithm takes into account data uncertainties via Poisson statistics modified by a noise contribution that is determined automatically during an initial normalization stage. Protein quantification relies on assignments of component peptides to the acquired data. These assignments are generally of variable reliability and may not be present across all of the experiments comprising an analysis. It is also possible for a peptide to be identified to more than one protein in a given mixture. For these reasons the algorithm accepts a prior probability of peptide assignment for each intensity measurement. The model is constructed in such a way that outliers of any type can be automatically reweighted. Two discrete normalization methods can be employed. The first method is based on a user-defined subset of peptides, while the second method relies on the presence of a dominant background of endogenous peptides for which the concentration is assumed to be unaffected. Normalization is performed using the same computational and statistical procedures employed by the main quantification algorithm. The performance of the algorithm will be illustrated on example data sets, and its utility demonstrated for typical proteomics applications. The quantification algorithm supports relative protein quantification based on precursor and product ion intensities acquired by means of data-dependent methods, originating from all common isotopically-labeled approaches, as well as label-free ion intensity-based data-independent methods.
Figures
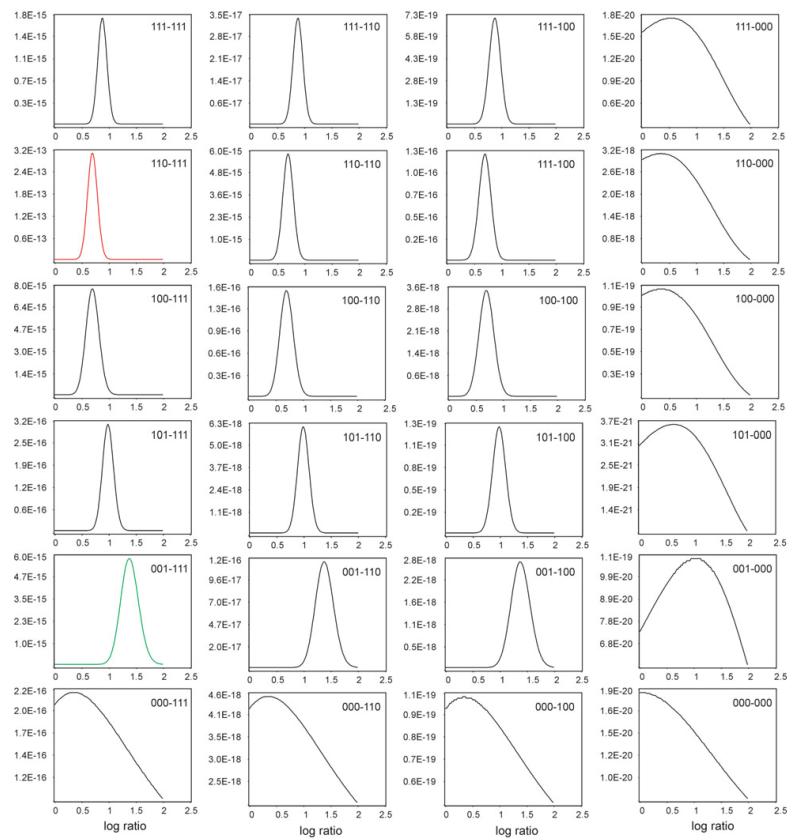
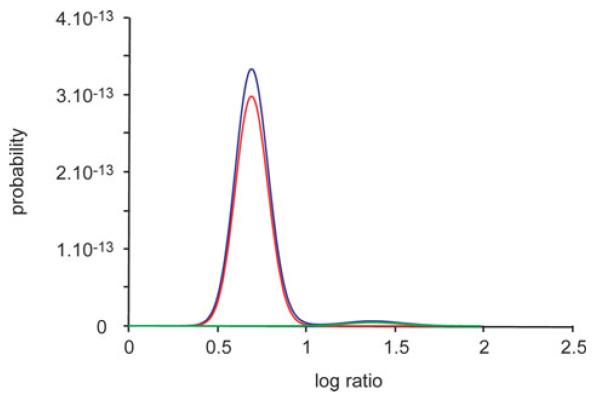
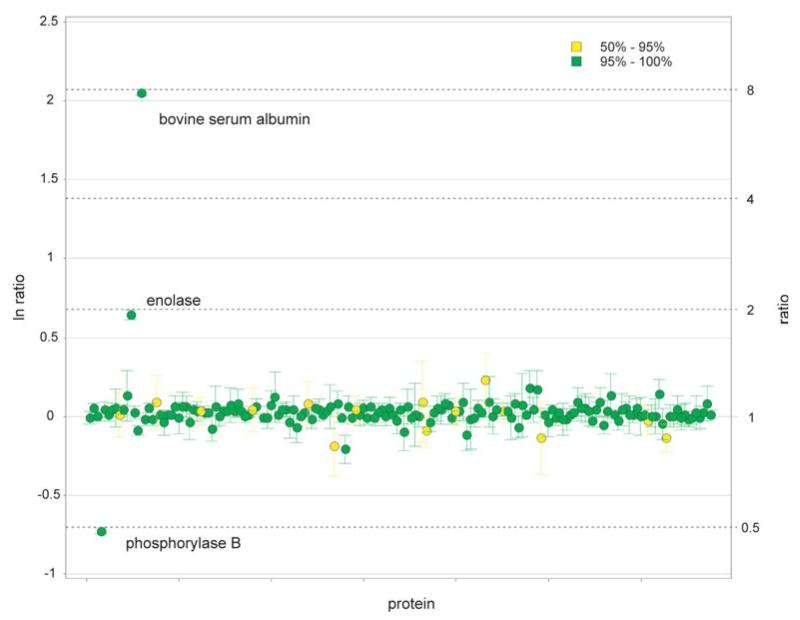
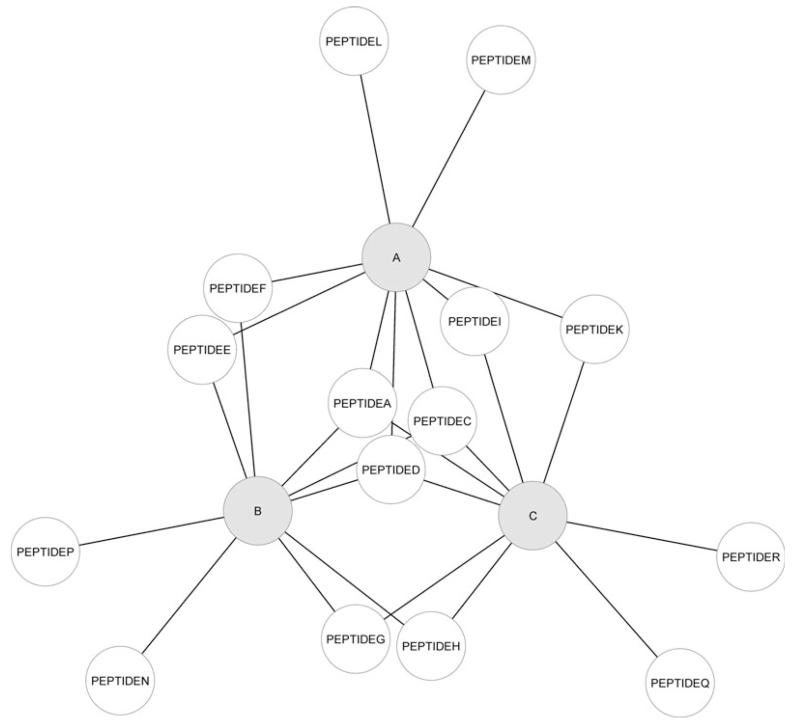
Similar articles
-
A statistical selection strategy for normalization procedures in LC-MS proteomics experiments through dataset-dependent ranking of normalization scaling factors.Proteomics. 2011 Dec;11(24):4736-41. doi: 10.1002/pmic.201100078. Epub 2011 Nov 17. Proteomics. 2011. PMID: 22038874 Free PMC article.
-
Corra: Computational framework and tools for LC-MS discovery and targeted mass spectrometry-based proteomics.BMC Bioinformatics. 2008 Dec 16;9:542. doi: 10.1186/1471-2105-9-542. BMC Bioinformatics. 2008. PMID: 19087345 Free PMC article.
-
Experimental design and data-analysis in label-free quantitative LC/MS proteomics: A tutorial with MSqRob.J Proteomics. 2018 Jan 16;171:23-36. doi: 10.1016/j.jprot.2017.04.004. Epub 2017 Apr 5. J Proteomics. 2018. PMID: 28391044
-
A comparative analysis of computational approaches to relative protein quantification using peptide peak intensities in label-free LC-MS proteomics experiments.Proteomics. 2013 Feb;13(3-4):493-503. doi: 10.1002/pmic.201200269. Epub 2012 Nov 8. Proteomics. 2013. PMID: 23019139 Free PMC article. Review.
-
Computational methods for the comparative quantification of proteins in label-free LCn-MS experiments.Brief Bioinform. 2008 Mar;9(2):156-65. doi: 10.1093/bib/bbm046. Epub 2007 Sep 28. Brief Bioinform. 2008. PMID: 17905794 Review.
Cited by
-
A comprehensive LFQ benchmark dataset on modern day acquisition strategies in proteomics.Sci Data. 2022 Mar 30;9(1):126. doi: 10.1038/s41597-022-01216-6. Sci Data. 2022. PMID: 35354825 Free PMC article.
-
Specificity of the osmotic stress response in Candida albicans highlighted by quantitative proteomics.Sci Rep. 2018 Sep 27;8(1):14492. doi: 10.1038/s41598-018-32792-6. Sci Rep. 2018. PMID: 30262823 Free PMC article.
-
Bioinformatics challenges and solutions in proteomics as quantitative methods mature.OMICS. 2012 Sep;16(9):429-30. doi: 10.1089/omi.2012.0051. Epub 2012 Jul 17. OMICS. 2012. PMID: 22804490 Free PMC article. No abstract available.
-
Scanning Quadrupole Data-Independent Acquisition, Part A: Qualitative and Quantitative Characterization.J Proteome Res. 2018 Feb 2;17(2):770-779. doi: 10.1021/acs.jproteome.7b00464. Epub 2017 Dec 29. J Proteome Res. 2018. PMID: 28901143 Free PMC article.
-
OLFM4, KNG1 and Sec24C identified by proteomics and immunohistochemistry as potential markers of early colorectal cancer stages.Clin Proteomics. 2017 Mar 24;14:9. doi: 10.1186/s12014-017-9143-3. eCollection 2017. Clin Proteomics. 2017. PMID: 28344541 Free PMC article.
References
-
- Aggarwal K, Choe LH, Lee KH. Quantitative analysis of protein expression using amine-specific isobaric tags in Escherichia coli cells expressing rhsA elements. Proteomics. 2005;5:2297–2308. - PubMed
-
- Bantscheff M, Schirle M, Sweetman G, Rick J, Kuster B. Quantitative mass spectrometry in proteomics: a critical review. Anal Bioanal Chem. 2007;389:1017–1031. - PubMed
-
- Bateman RH, Carruthers R, Hoyes JB, et al. A novel precursor ion discovery method on a hybrid quadrupole orthogonal acceleration time-of-flight (Q-TOF) mass spectrometer for studying protein phosphorylation. J Am Soc Mass Spectrom. 2002;13:792–803. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
