

Genotoxic stress modulates CDC25C phosphatase alternative splicing in human breast cancer cell lines
- PMID: 22871320
- PMCID: PMC5528395
- DOI: 10.1016/j.molonc.2012.06.003
Genotoxic stress modulates CDC25C phosphatase alternative splicing in human breast cancer cell lines
Abstract
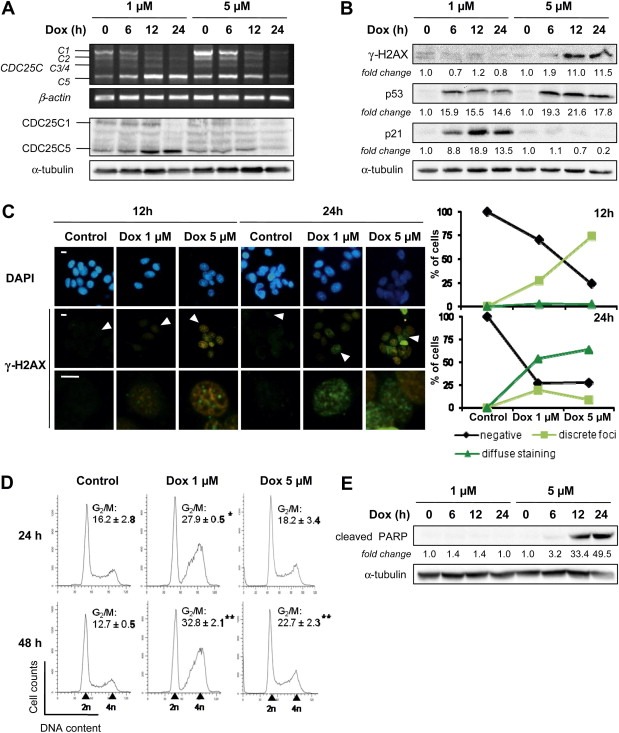
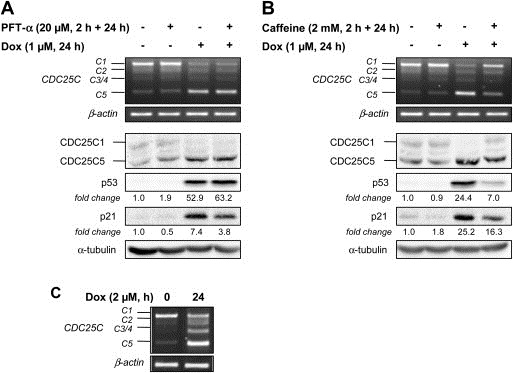
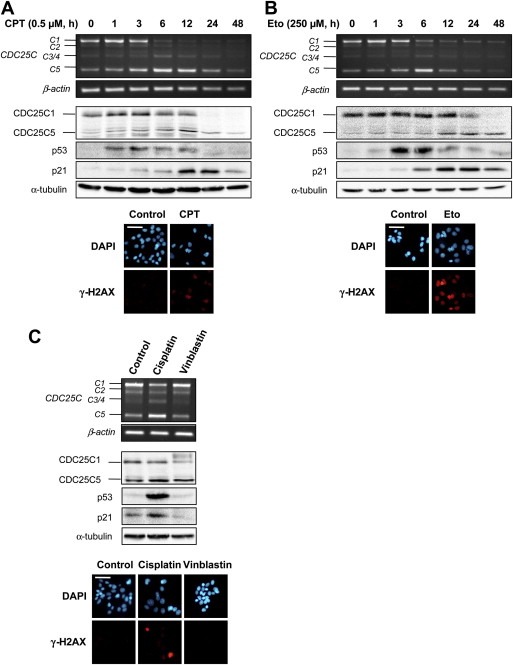
CDC25 (cell division cycle 25) phosphatases are essential for cell cycle control under normal conditions and in response to DNA damage. They are represented by three isoforms, CDC25A, B and C, each of them being submitted to an alternative splicing mechanism. Alternative splicing of many genes is affected in response to genotoxic stress, but the impact of such a stress on CDC25 splicing has never been investigated. In this study, we demonstrate that genotoxic agents (doxorubicin, camptothecin, etoposide and cisplatin), alter the balance between CDC25C splice variants in human breast cancer cell lines both at the mRNA and protein levels. This modulation occurs during the response to moderate, sub-lethal DNA damage. Our results also suggest that the CDC25C splice variants expression shift induced by a genotoxic stress is dependent on the ATM/ATR signaling but not on p53. This study highlights the modulation of CDC25C alternative splicing as an additional regulatory event involved in cellular response to DNA damage in breast cancer cells.
Copyright © 2012 Federation of European Biochemical Societies. Published by Elsevier B.V. All rights reserved.
Figures

References
-
- Albert, H. , Santos, S. , Battaglia, E. , Brito, M. , Monteiro, C. , Bagrel, D. , 2011. Differential expression of CDC25 phosphatases splice variants in human breast cancer cells. Clin. Chem. Lab. Med.. 49, 1707–1714. - PubMed
-
- Aressy, B. , Ducommun, B. , 2008. Cell cycle control by the CDC25 phosphatases. Anticancer Agents Med. Chem.. 8, 818–824. - PubMed
-
- Baldin, V. , Cans, C. , Superti-Furga, G. , Ducommun, B. , 1997. Alternative splicing of the human CDC25B tyrosine phosphatase. Possible implications for growth control?. Oncogene. 14, 2485–2495. - PubMed
-
- Bartek, J. , Iggo, R. , Gannon, J. , Lane, D.P. , 1990. Genetic and immochemical analysis of mutant p53 in human breast cancer cell lines. Oncogene. 5, 893–899. - PubMed
-
- Biamonti, G. , Caceres, J.F. , 2009. Cellular stress and RNA splicing. Trends Biochem. Sci.. 34, 146–153. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
