

KRAS(G12D)- and BRAF(V600E)-induced transformation of murine pancreatic epithelial cells requires MEK/ERK-stimulated IGF1R signaling
- PMID: 22871572
- PMCID: PMC3973739
- DOI: 10.1158/1541-7786.MCR-12-0340-T
KRAS(G12D)- and BRAF(V600E)-induced transformation of murine pancreatic epithelial cells requires MEK/ERK-stimulated IGF1R signaling
Abstract
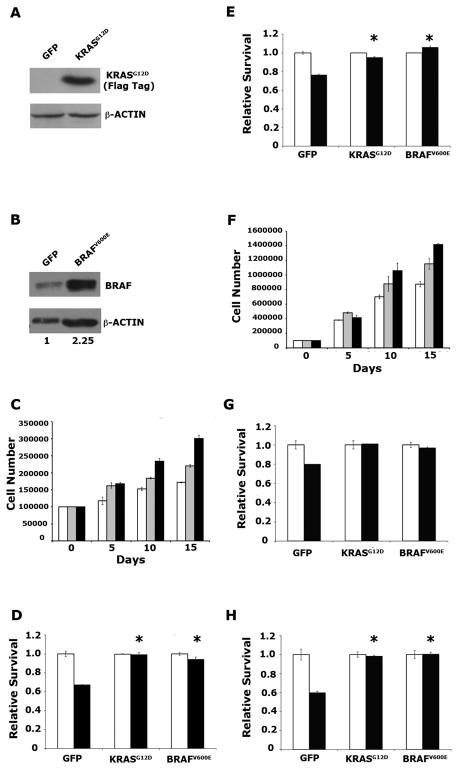
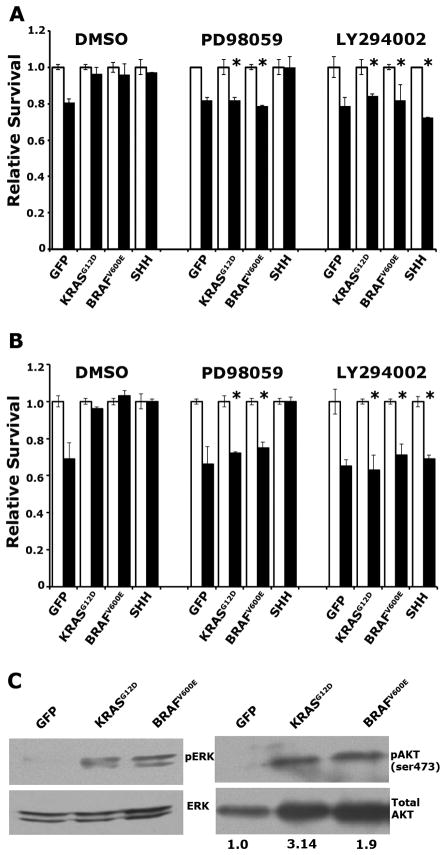
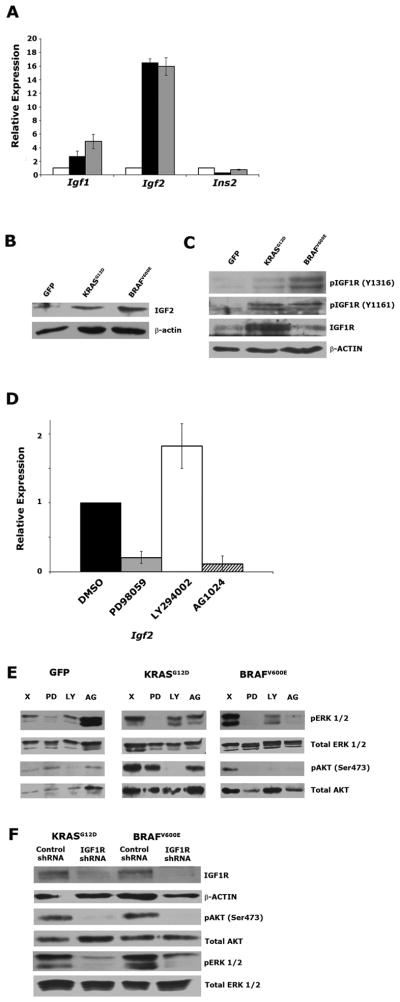
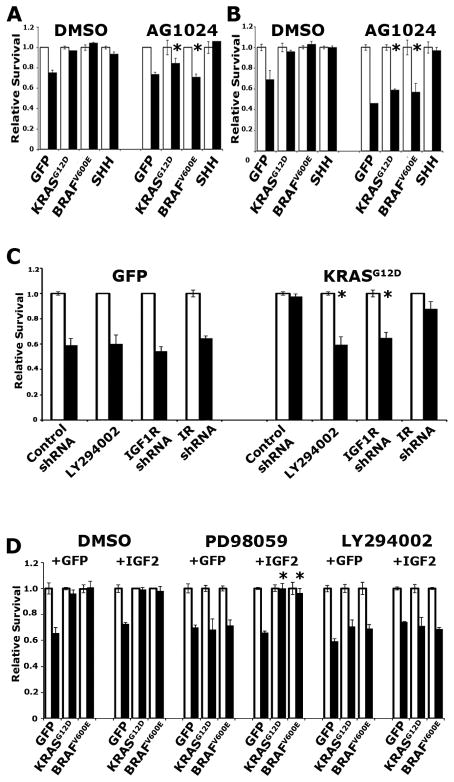
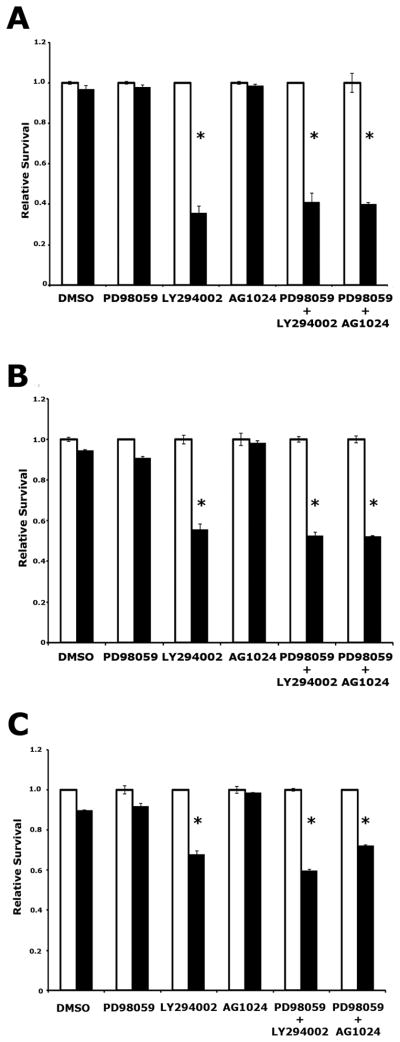
Mutation of KRAS is a common initiating event in pancreatic ductal adenocarcinoma (PDAC). Yet, the specific roles of KRAS-stimulated signaling pathways in the transformation of pancreatic ductal epithelial cells (PDEC), putative cells of origin for PDAC, remain unclear. Here, we show that KRAS(G12D) and BRAF(V600E) enhance PDEC proliferation and increase survival after exposure to apoptotic stimuli in a manner dependent on MEK/ERK and PI3K/AKT signaling. Interestingly, we find that activation of PI3K/AKT signaling occurs downstream of MAP-ERK kinase (MEK), and is dependent on the autocrine activation of the insulin-like growth factor (IGF) receptor (IGF1R) by IGF2. Importantly, IGF1R inhibition impairs KRAS(G12D)- and BRAF(V600E)-induced survival, whereas ectopic IGF2 expression rescues KRAS(G12D)- and BRAF(V600E)-mediated survival downstream of MEK inhibition. Moreover, we show that KRAS(G12D)- and BRAF(V600E)-induced tumor formation in an orthotopic model requires IGF1R. Interestingly, we show that while individual inhibition of MEK or IGF1R does not sensitize PDAC cells to apoptosis, their concomitant inhibition reduces survival. Our findings identify a novel mechanism of PI3K/AKT activation downstream of activated KRAS, illustrate the importance of MEK/ERK, PI3K/AKT, and IGF1R signaling in pancreatic tumor initiation, and suggest potential therapeutic strategies for this malignancy.
Figures
Similar articles
-
Loss of Somatostatin Receptor Subtype 2 Promotes Growth of KRAS-Induced Pancreatic Tumors in Mice by Activating PI3K Signaling and Overexpression of CXCL16.Gastroenterology. 2015 Jun;148(7):1452-65. doi: 10.1053/j.gastro.2015.02.009. Epub 2015 Feb 13. Gastroenterology. 2015. PMID: 25683115
-
The suppressive efficacy of THZ1 depends on KRAS mutation subtype and is associated with super-enhancer activity and the PI3K/AKT/mTOR signalling in pancreatic ductal adenocarcinoma: A hypothesis-generating study.Clin Transl Med. 2023 Dec;13(12):e1500. doi: 10.1002/ctm2.1500. Clin Transl Med. 2023. PMID: 38037549 Free PMC article.
-
Nicotine promotes initiation and progression of KRAS-induced pancreatic cancer via Gata6-dependent dedifferentiation of acinar cells in mice.Gastroenterology. 2014 Nov;147(5):1119-33.e4. doi: 10.1053/j.gastro.2014.08.002. Epub 2014 Aug 12. Gastroenterology. 2014. PMID: 25127677
-
Inhibition of the RAF/MEK/ERK Signaling Cascade in Pancreatic Cancer: Recent Advances and Future Perspectives.Int J Mol Sci. 2024 Jan 28;25(3):1631. doi: 10.3390/ijms25031631. Int J Mol Sci. 2024. PMID: 38338909 Free PMC article. Review.
-
KRAS Pathway-based Therapeutic Approaches in Pancreatic Cancer.Mini Rev Med Chem. 2023;23(8):953-961. doi: 10.2174/1389557523666221226095931. Mini Rev Med Chem. 2023. PMID: 36573057 Review.
Cited by
-
Effects of insulin on human pancreatic cancer progression modeled in vitro.BMC Cancer. 2014 Nov 6;14:814. doi: 10.1186/1471-2407-14-814. BMC Cancer. 2014. PMID: 25373319 Free PMC article.
-
Remarkable response of BRAF V600E-mutated metastatic pancreatic cancer to BRAF/MEK inhibition: a case report.Gastroenterol Rep (Oxf). 2021 Sep 13;10:goab031. doi: 10.1093/gastro/goab031. eCollection 2022. Gastroenterol Rep (Oxf). 2021. PMID: 35382161 Free PMC article. No abstract available.
-
Pancreatic endocrine and exocrine signaling and crosstalk in physiological and pathological status.Signal Transduct Target Ther. 2025 Feb 14;10(1):39. doi: 10.1038/s41392-024-02098-3. Signal Transduct Target Ther. 2025. PMID: 39948335 Free PMC article. Review.
-
Kras(G12D) induces EGFR-MYC cross signaling in murine primary pancreatic ductal epithelial cells.Oncogene. 2016 Jul 21;35(29):3880-6. doi: 10.1038/onc.2015.437. Epub 2015 Nov 23. Oncogene. 2016. PMID: 26592448 Free PMC article.
-
Obesity and endocrine-related cancer: The important role of IGF-1.Front Endocrinol (Lausanne). 2023 Jan 23;14:1093257. doi: 10.3389/fendo.2023.1093257. eCollection 2023. Front Endocrinol (Lausanne). 2023. PMID: 36755926 Free PMC article. Review.
References
-
- Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA: a cancer journal for clinicians. 2010;60:277–300. - PubMed
-
- Hruban RH, Goggins M, Parsons J, Kern SE. Progression model for pancreatic cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2000;6:2969–72. - PubMed
-
- Bardeesy N, DePinho RA. Pancreatic cancer biology and genetics. Nature reviews Cancer. 2002;2:897–909. - PubMed
-
- Hezel AF, Kimmelman AC, Stanger BZ, Bardeesy N, Depinho RA. Genetics and biology of pancreatic ductal adenocarcinoma. Genes & development. 2006;20:1218–49. - PubMed
-
- Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53:549–54. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
