

Microtubules underlie dysfunction in duchenne muscular dystrophy
- PMID: 22871609
- PMCID: PMC3835660
- DOI: 10.1126/scisignal.2002829
Microtubules underlie dysfunction in duchenne muscular dystrophy
Abstract
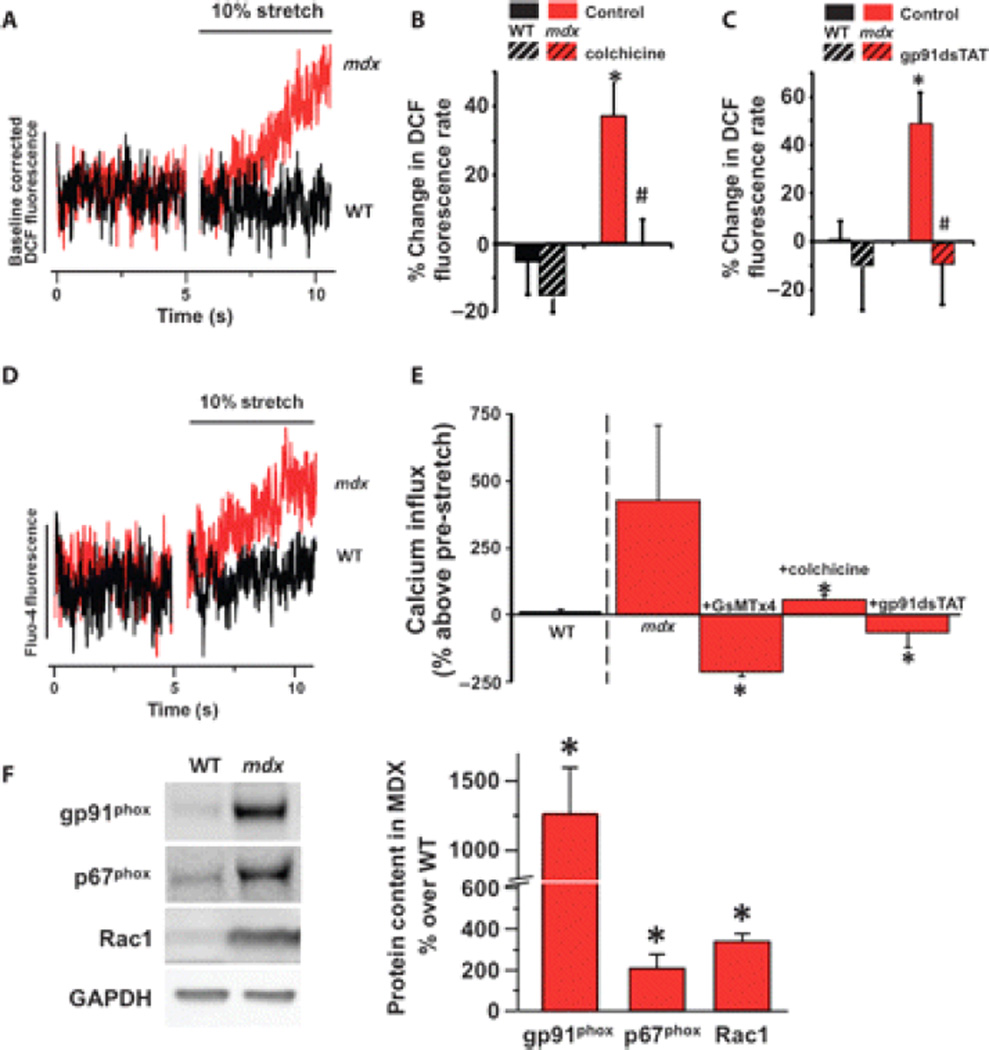
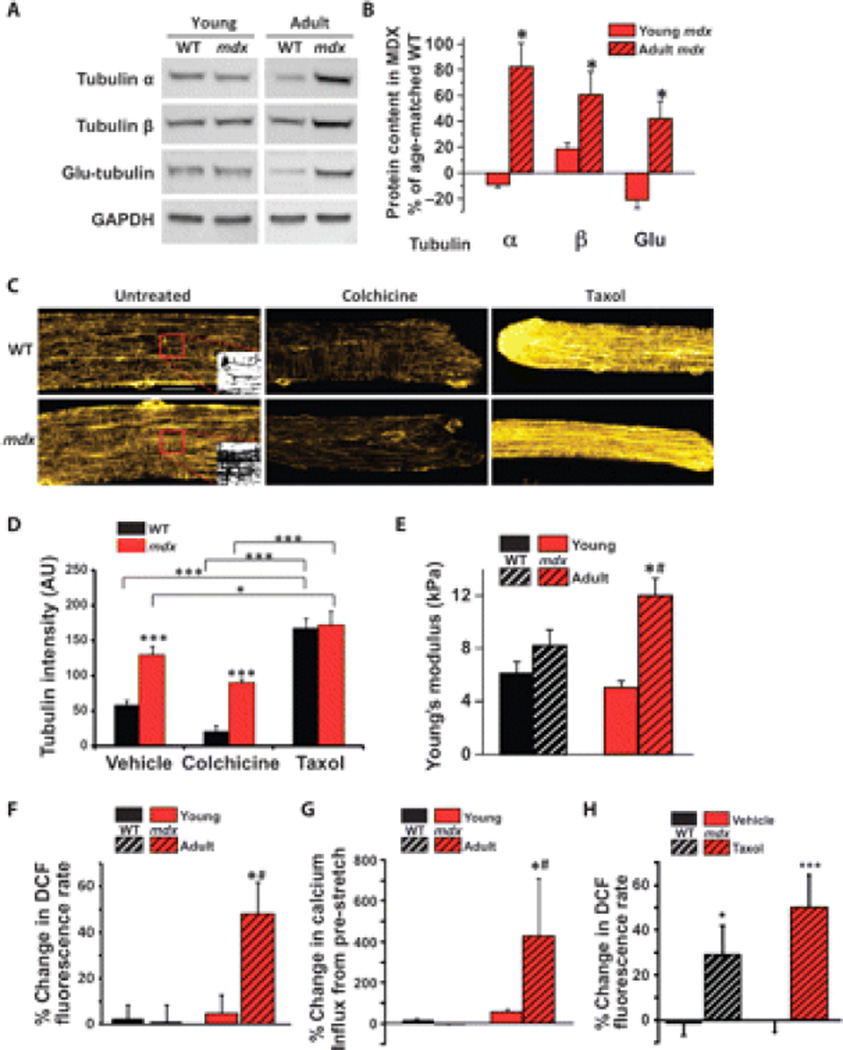
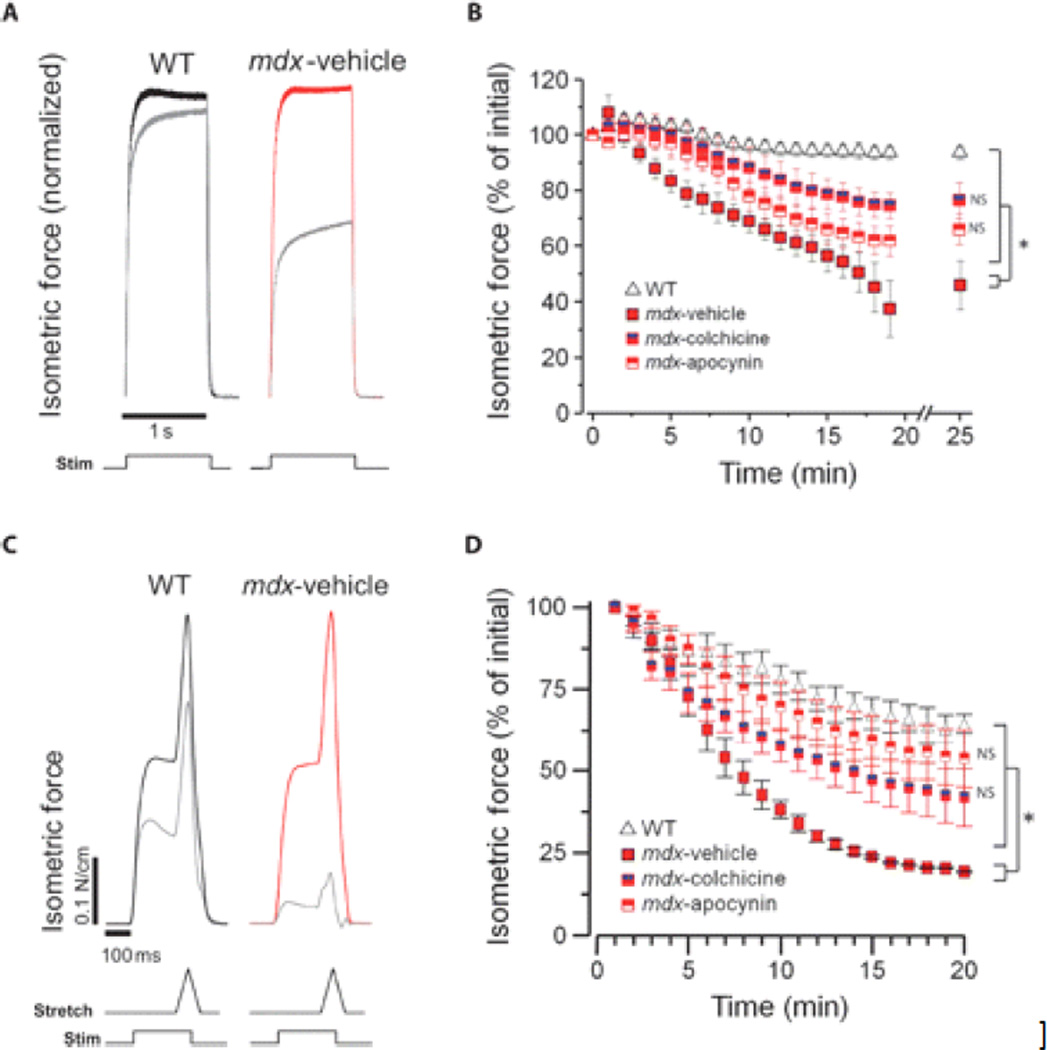
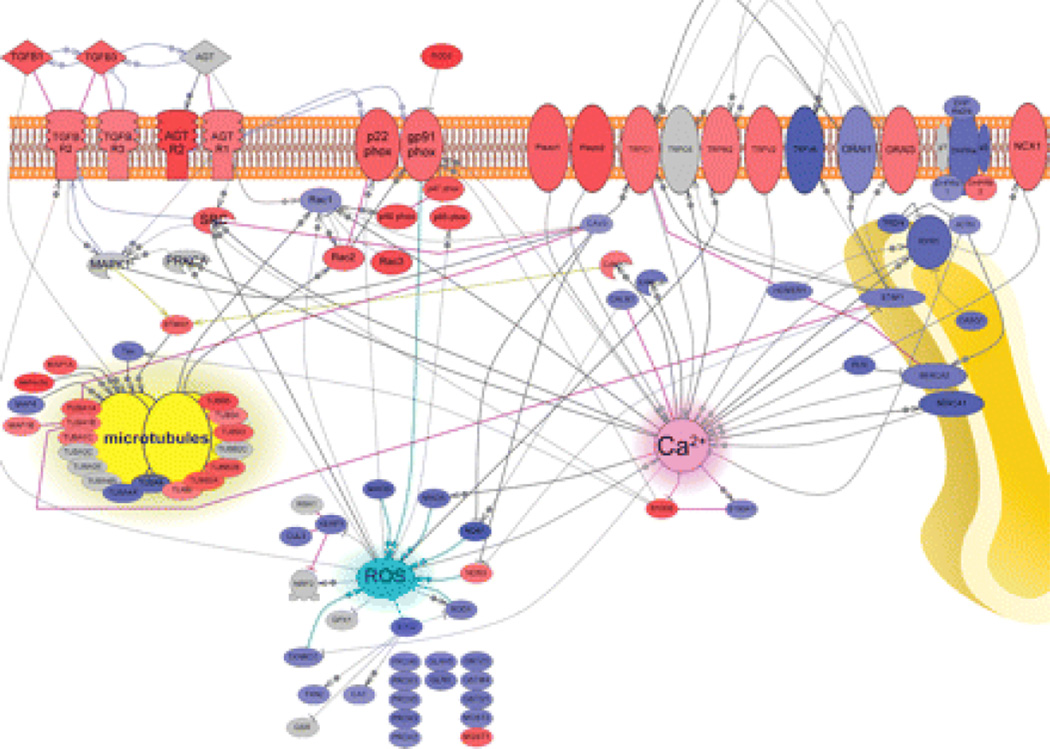
Duchenne muscular dystrophy (DMD) is a fatal X-linked degenerative muscle disease caused by the absence of the microtubule-associated protein dystrophin, which results in a disorganized and denser microtubule cytoskeleton. In addition, mechanotransduction-dependent activation of calcium (Ca(2+)) and reactive oxygen species (ROS) signaling underpins muscle degeneration in DMD. We show that in muscle from adult mdx mice, a model of DMD, a brief physiologic stretch elicited microtubule-dependent activation of NADPH (reduced-form nicotinamide adenine dinucleotide phosphate) oxidase-dependent production of ROS, termed X-ROS. Further, X-ROS amplified Ca(2+) influx through stretch-activated channels in mdx muscle. Consistent with the importance of the microtubules to the dysfunction in mdx muscle, muscle cells with dense microtubule structure, such as those from adult mdx mice or from young wild-type mice treated with Taxol, showed increased X-ROS production and Ca(2+) influx, whereas cells with a less dense microtubule network, such as young mdx or adult mdx muscle treated with colchicine or nocodazole, showed little ROS production or Ca(2+) influx. In vivo treatments that disrupted the microtubule network or inhibited NADPH oxidase 2 reduced contraction-induced injury in adult mdx mice. Furthermore, transcriptome analysis identified increased expression of X-ROS-related genes in human DMD skeletal muscle. Together, these data show that microtubules are the proximate element responsible for the dysfunction in Ca(2+) and ROS signaling in DMD and could be effective therapeutic targets for intervention.
Figures
References
-
- Hoffman EP, Brown RH, Jr, Kunkel LM. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. - PubMed
-
- Pegoraro E, Hoffman EP, Piva L, Gavassini BF, Cagnin S, Ermani M, Bello L, Soraru G, Pacchioni B, Bonifati MD, Lanfranchi G, Angelini C, Kesari A, Lee I, Gordish-Dressman H, Devaney JM, McDonald CM. SPP1 genotype is a determinant of disease severity in Duchenne muscular dystrophy. Neurology. 2011;76:219–226. - PMC - PubMed
-
- Allen DG, Gervasio OL, Yeung EW, Whitehead NP. Calcium and the damage pathways in muscular dystrophy. Can. J. Physiol. Pharmacol. 2010;88:83–91. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- U54HD053177/HD/NICHD NIH HHS/United States
- T32 AR007592/AR/NIAMS NIH HHS/United States
- R01 HL36974/HL/NHLBI NIH HHS/United States
- R01 NS029525/NS/NINDS NIH HHS/United States
- R01 HL036974/HL/NHLBI NIH HHS/United States
- U54 HD053177/HD/NICHD NIH HHS/United States
- P01 HL067849/HL/NHLBI NIH HHS/United States
- R01 HL106059/HL/NHLBI NIH HHS/United States
- R03 TW008425/TW/FIC NIH HHS/United States
- R01 HL105239/HL/NHLBI NIH HHS/United States
- R01 AR062554/AR/NIAMS NIH HHS/United States
- L40 AR056534/AR/NIAMS NIH HHS/United States
- T32 HL072751/HL/NHLBI NIH HHS/United States
- RC2 NR011968/NR/NINR NIH HHS/United States
- T32AR007592/AR/NIAMS NIH HHS/United States
- P01 HL67849/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
