

Impaired proteolysis underlies autophagic dysfunction in Niemann-Pick type C disease
- PMID: 22872701
- PMCID: PMC3607480
- DOI: 10.1093/hmg/dds324
Impaired proteolysis underlies autophagic dysfunction in Niemann-Pick type C disease
Abstract
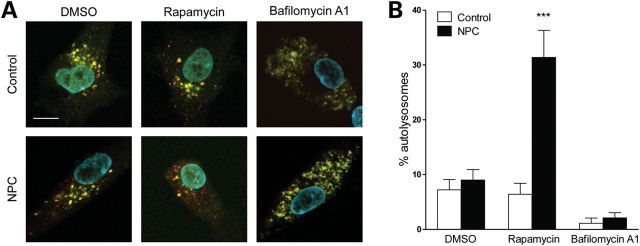
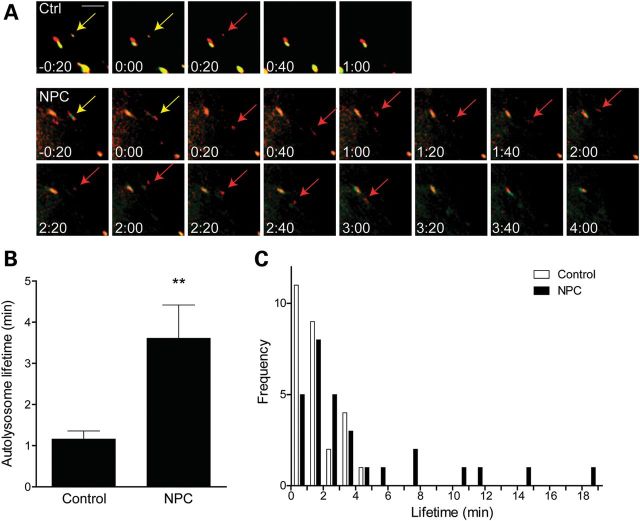
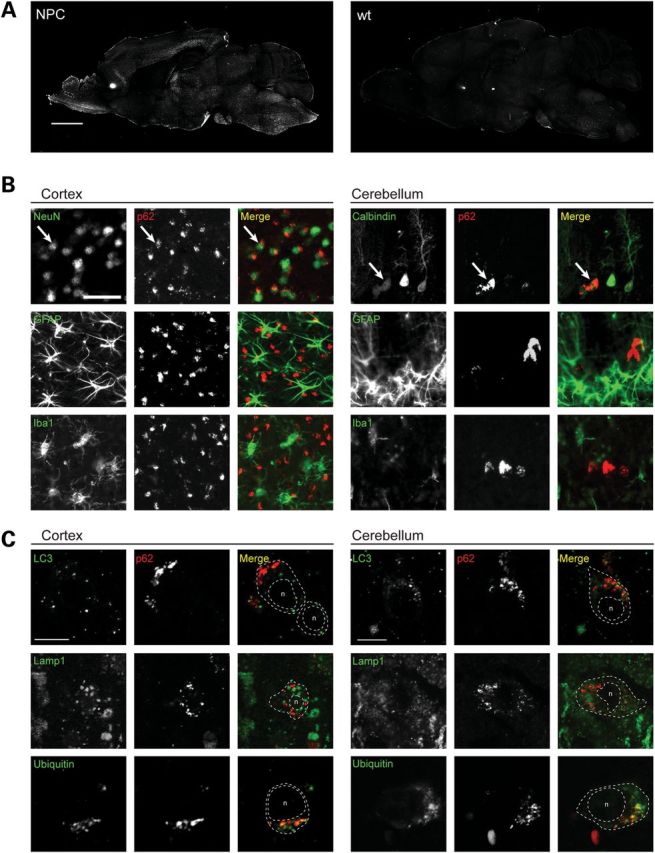
Niemann-Pick type C disease (NPC) is a childhood onset neurodegenerative disorder arising from lipid-trafficking defects caused by mutations in the NPC1 or NPC2 gene. Marked accumulation of autophagosomes is a prominent feature of NPC cells, yet a detailed understanding of the disease-associated alterations in autophagy and their role in pathogenesis has been lacking. Prior studies have shown that lipid storage in NPC disease induces autophagy. Here, we additionally show that the clearance of autophagosomes in NPC1 deficiency is impaired due to inhibition of lysosomal protease activity by stored lipids. We also demonstrate that the autophagic pathway is a source of stored cholesterol in the NPC lysosome, thus creating a positive feedback loop wherein autophagy induction exacerbates the disease via increased lipid storage. Inhibition of autophagy reduces cholesterol storage and restores normal lysosomal proteolysis in NPC1-deficient cells, supporting a model in which activation of the autophagic pathway promotes disease pathogenesis.
Figures




References
-
- Hara T., Nakamura K., Matsui M., Yamamoto A., Nakahara Y., Suzuki-Migishima R., Yokoyama M., Mishima K., Saito I., Okano H., et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. - PubMed
-
- Komatsu M., Waguri S., Chiba T., Murata S., Iwata J., Tanida I., Ueno T., Koike M., Uchiyama Y., Kominami E., et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. - PubMed
-
- Ravikumar B., Vacher C., Berger Z., Davies J.E., Luo S., Oroz L.G., Scaravilli F., Easton D.F., Duden R., O'Kane C.J., et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004;36:585–595. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
