

Metabolic reprogramming of cancer-associated fibroblasts by TGF-β drives tumor growth: connecting TGF-β signaling with "Warburg-like" cancer metabolism and L-lactate production
- PMID: 22874531
- PMCID: PMC3442913
- DOI: 10.4161/cc.21384
Metabolic reprogramming of cancer-associated fibroblasts by TGF-β drives tumor growth: connecting TGF-β signaling with "Warburg-like" cancer metabolism and L-lactate production
Abstract
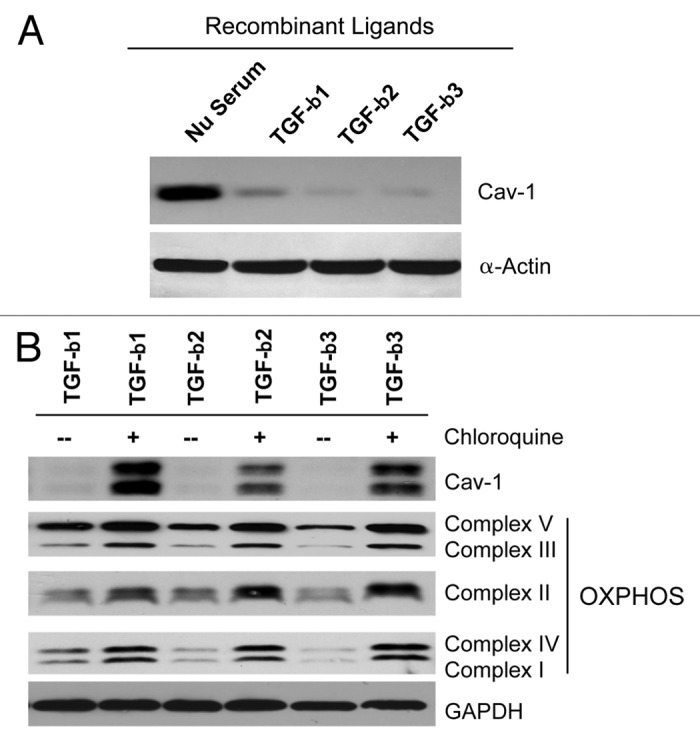
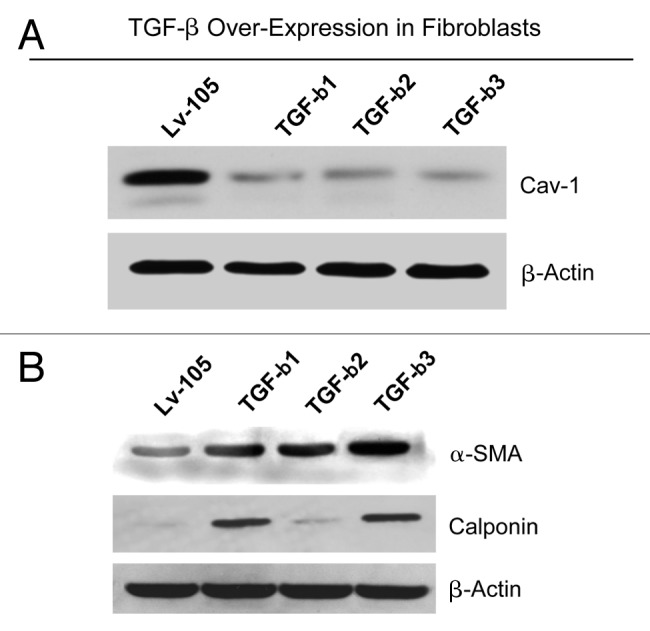
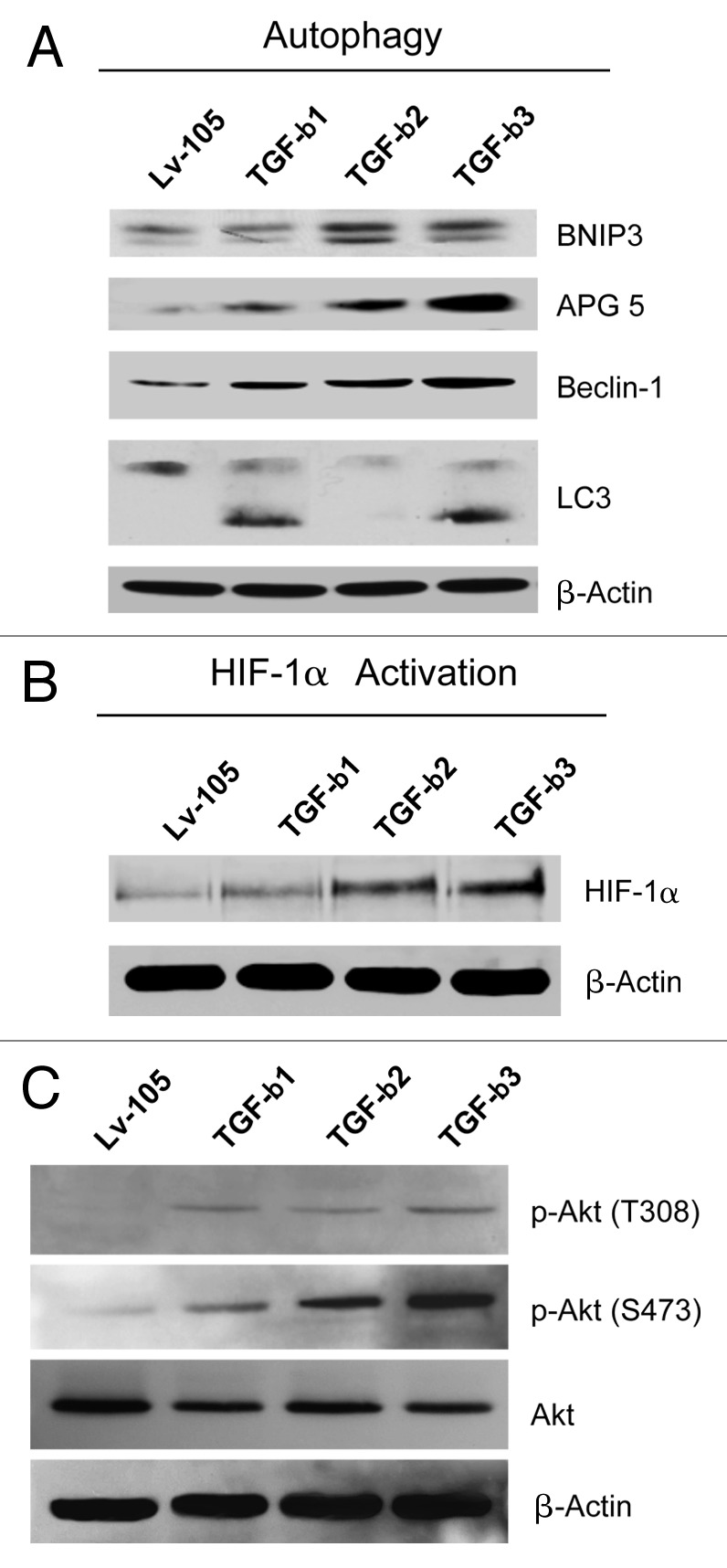
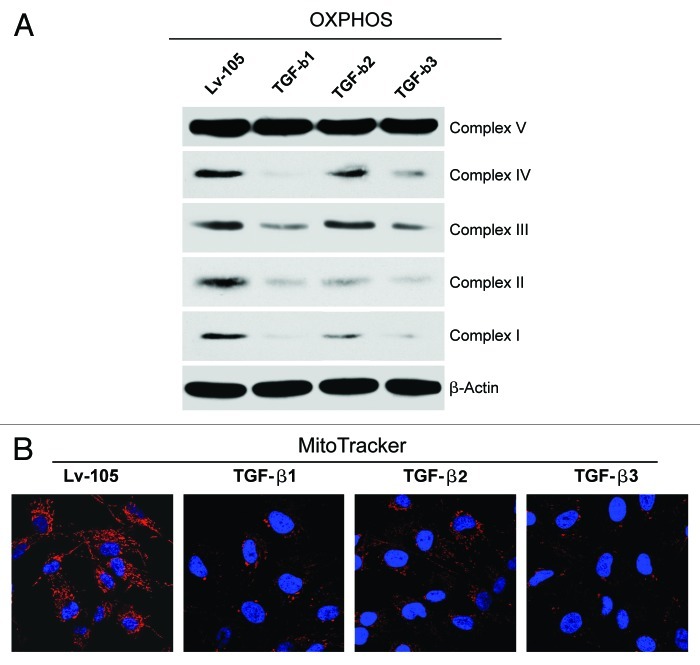
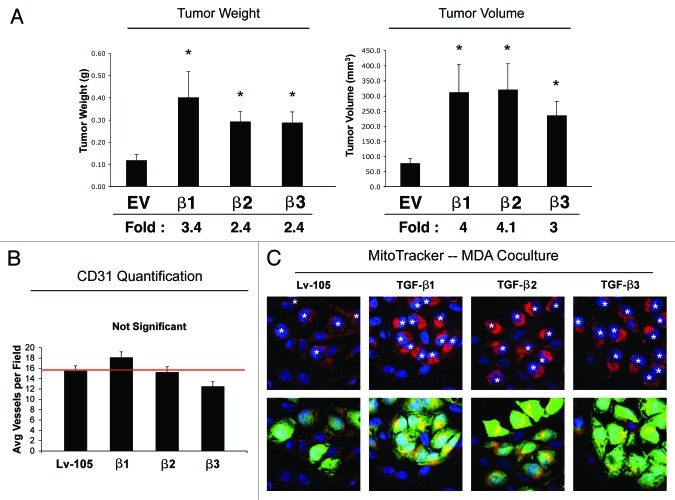
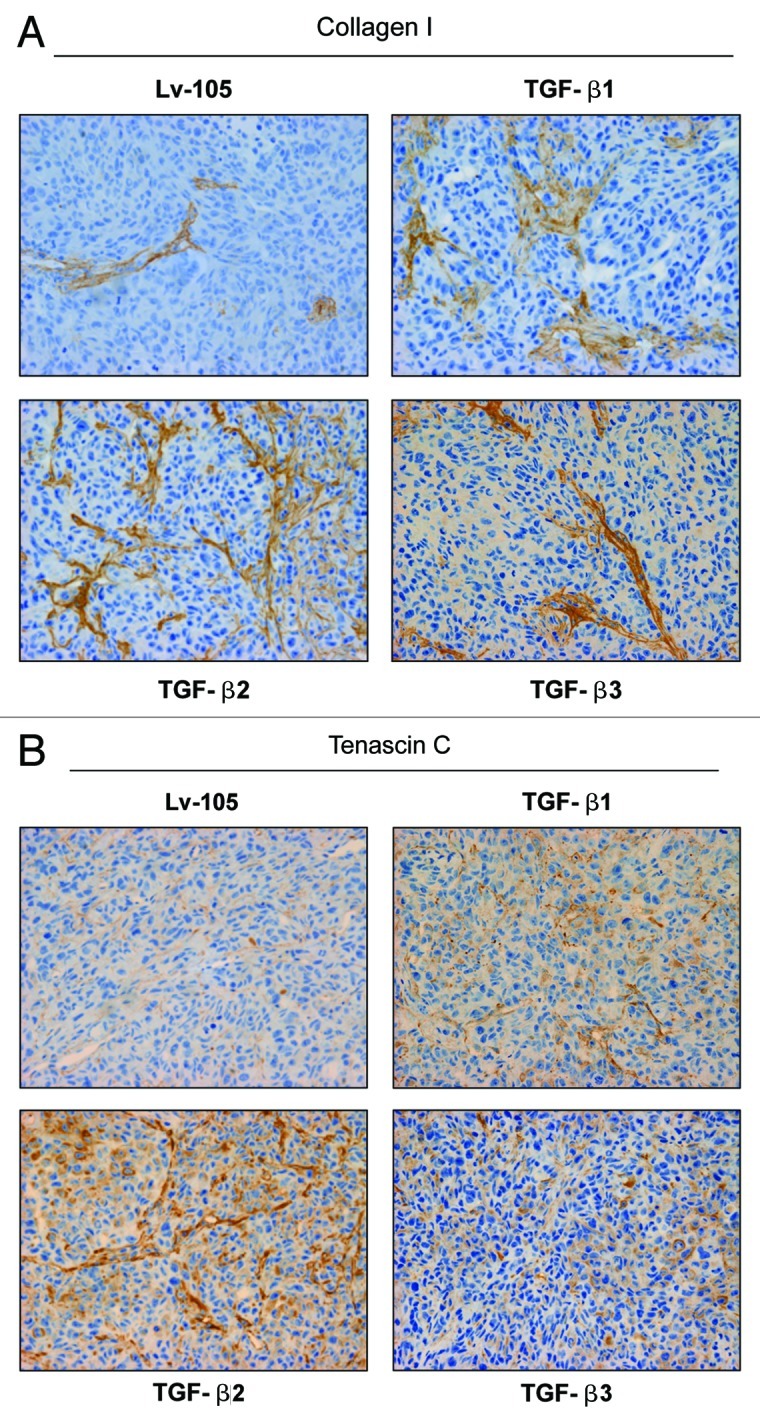
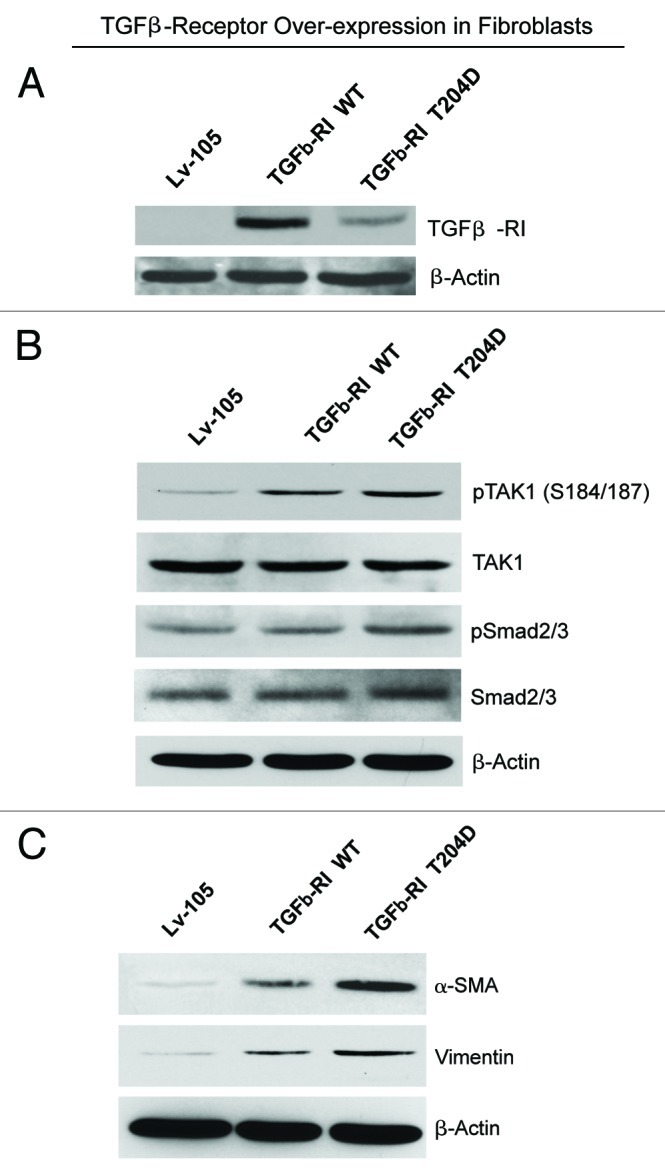
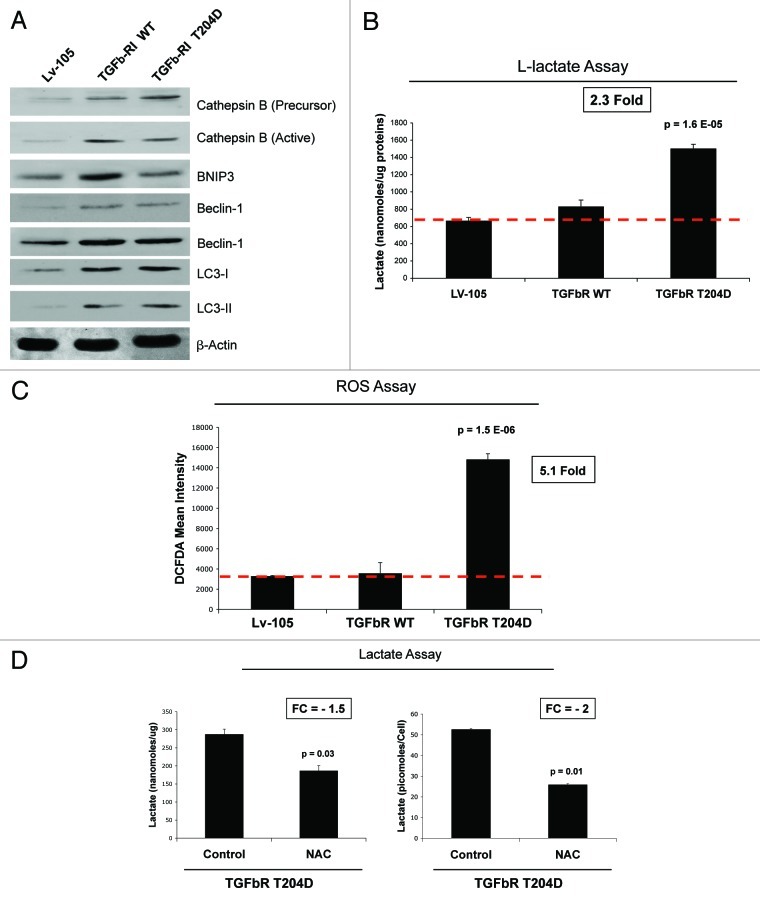
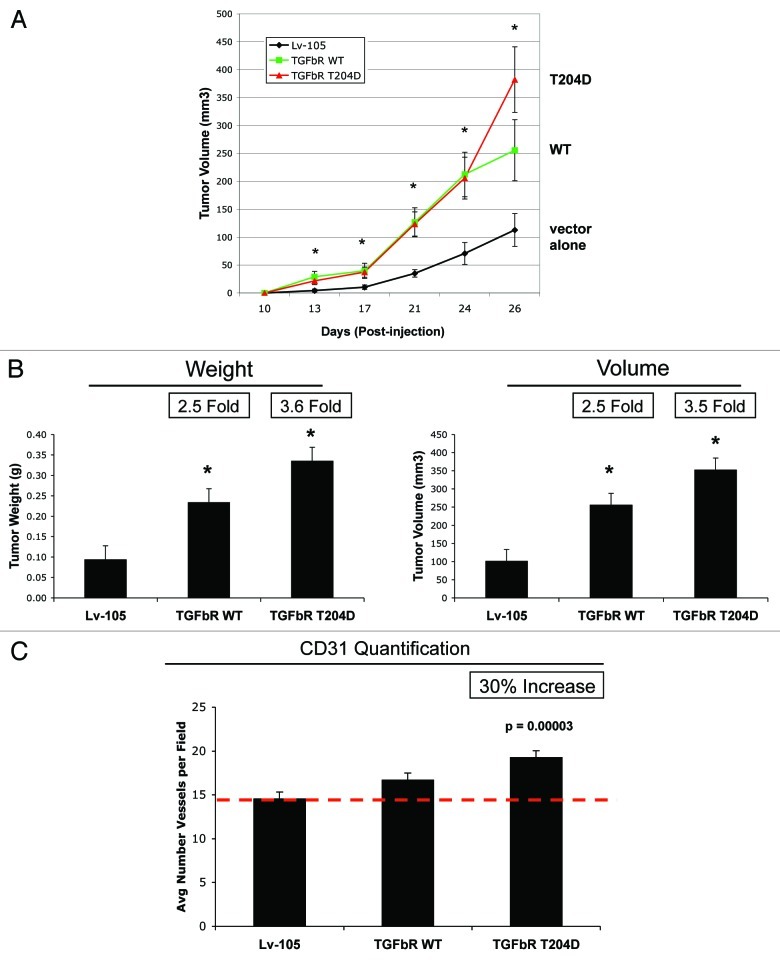
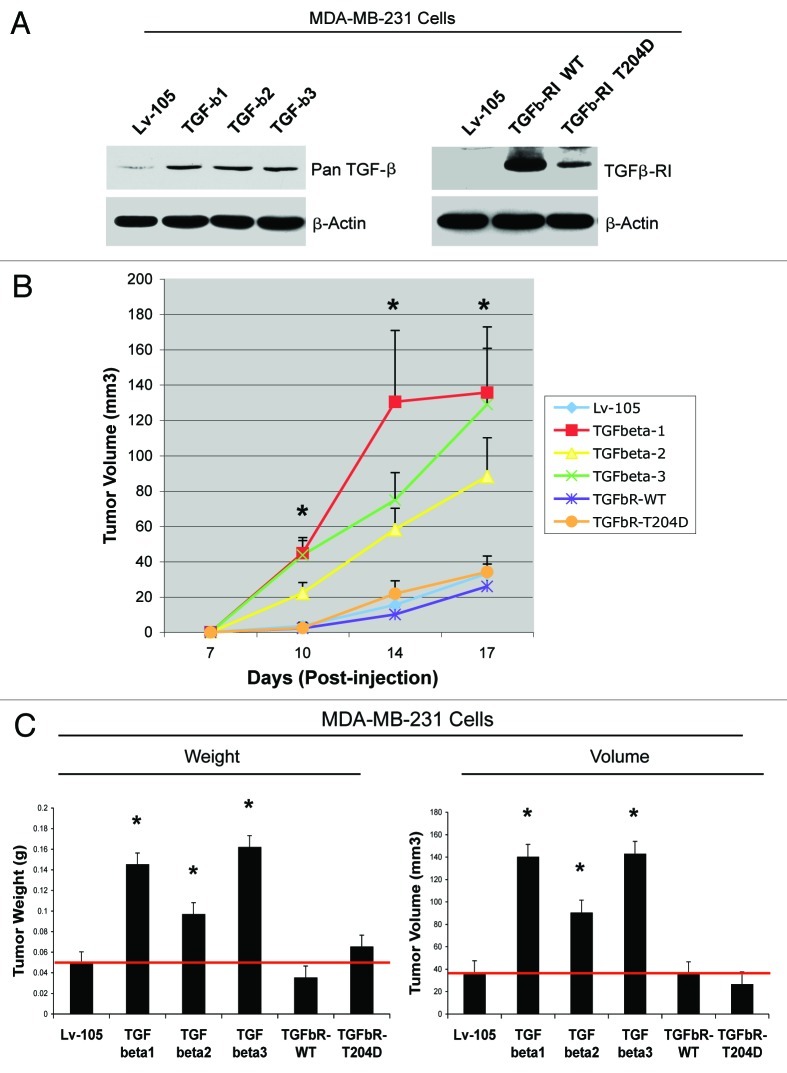
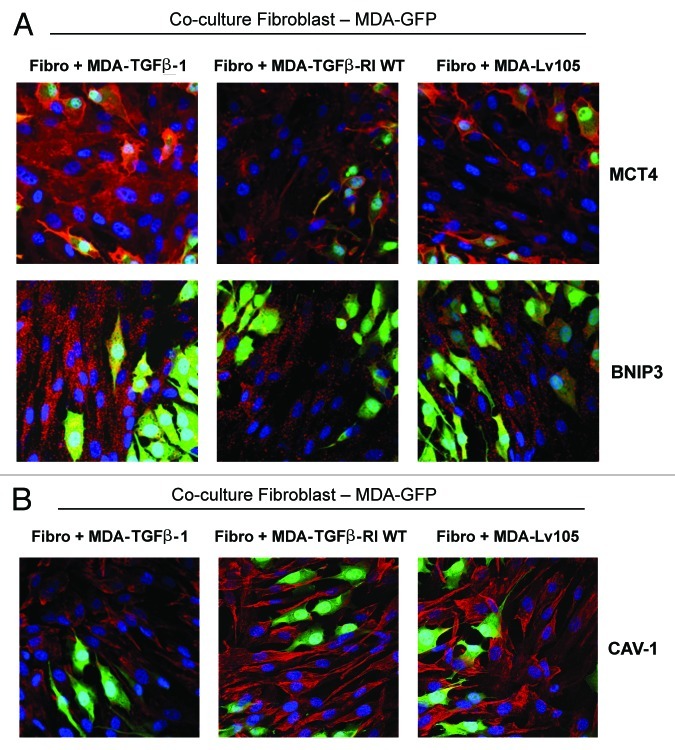
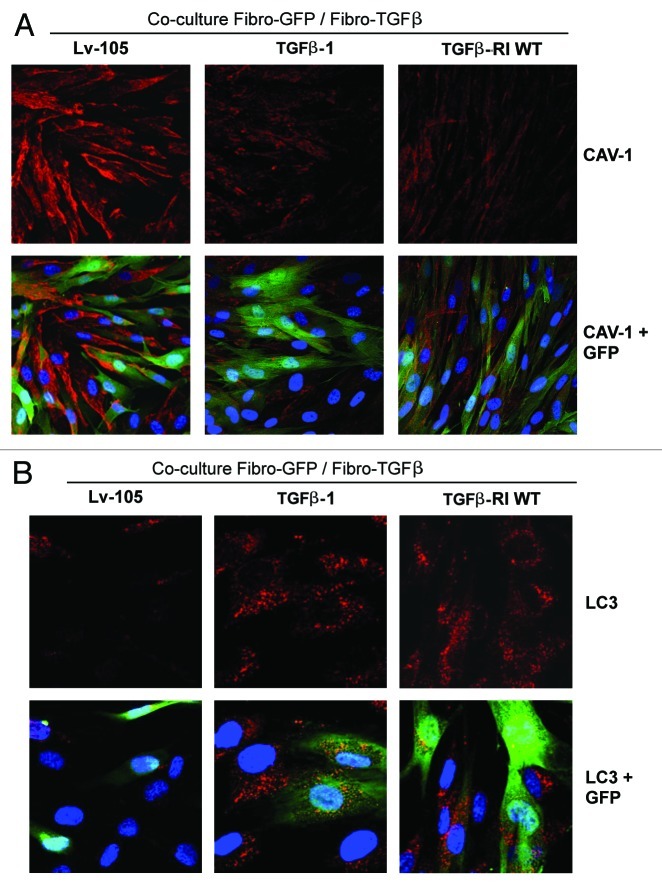
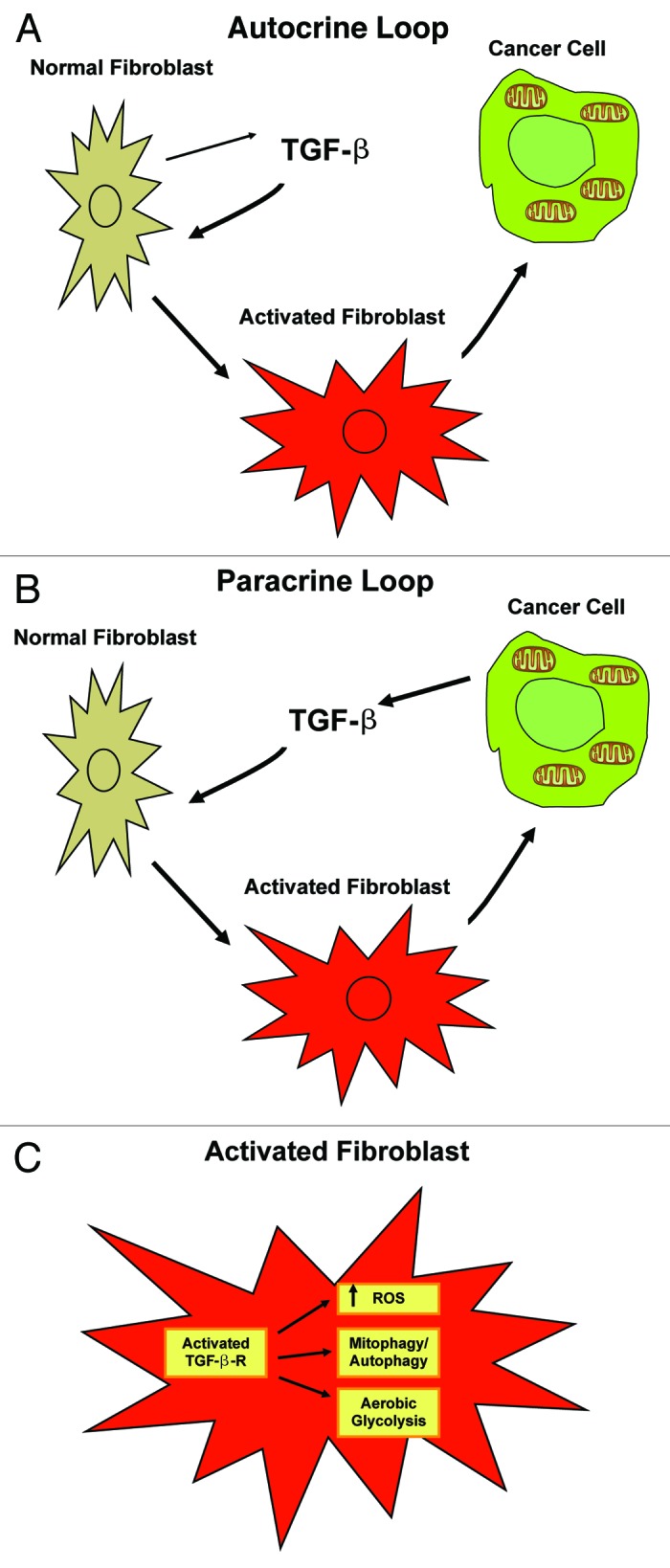
We have previously shown that a loss of stromal Cav-1 is a biomarker of poor prognosis in breast cancers. Mechanistically, a loss of Cav-1 induces the metabolic reprogramming of stromal cells, with increased autophagy/mitophagy, mitochondrial dysfunction and aerobic glycolysis. As a consequence, Cav-1-low CAFs generate nutrients (such as L-lactate) and chemical building blocks that fuel mitochondrial metabolism and the anabolic growth of adjacent breast cancer cells. It is also known that a loss of Cav-1 is associated with hyperactive TGF-β signaling. However, it remains unknown whether hyperactivation of the TGF-β signaling pathway contributes to the metabolic reprogramming of Cav-1-low CAFs. To address these issues, we overexpressed TGF-β ligands and the TGF-β receptor I (TGFβ-RI) in stromal fibroblasts and breast cancer cells. Here, we show that the role of TGF-β in tumorigenesis is compartment-specific, and that TGF-β promotes tumorigenesis by shifting cancer-associated fibroblasts toward catabolic metabolism. Importantly, the tumor-promoting effects of TGF-β are independent of the cell type generating TGF-β. Thus, stromal-derived TGF-β activates signaling in stromal cells in an autocrine fashion, leading to fibroblast activation, as judged by increased expression of myofibroblast markers, and metabolic reprogramming, with a shift toward catabolic metabolism and oxidative stress. We also show that TGF-β-activated fibroblasts promote the mitochondrial activity of adjacent cancer cells, and in a xenograft model, enhancing the growth of breast cancer cells, independently of angiogenesis. Conversely, activation of the TGF-β pathway in cancer cells does not influence tumor growth, but cancer cell-derived-TGF-β ligands affect stromal cells in a paracrine fashion, leading to fibroblast activation and enhanced tumor growth. In conclusion, ligand-dependent or cell-autonomous activation of the TGF-β pathway in stromal cells induces their metabolic reprogramming, with increased oxidative stress, autophagy/mitophagy and glycolysis, and downregulation of Cav-1. These metabolic alterations can spread among neighboring fibroblasts and greatly sustain the growth of breast cancer cells. Our data provide novel insights into the role of the TGF-β pathway in breast tumorigenesis, and establish a clear causative link between the tumor-promoting effects of TGF-β signaling and the metabolic reprogramming of the tumor microenvironment.
Figures
Comment in
-
Transforming growth factor-β: guardian of catabolic metabolism in carcinoma-associated fibroblasts.Cell Cycle. 2012 Dec 1;11(23):4302-3. doi: 10.4161/cc.22811. Epub 2012 Nov 16. Cell Cycle. 2012. PMID: 23159856 Free PMC article. No abstract available.
Similar articles
-
CTGF drives autophagy, glycolysis and senescence in cancer-associated fibroblasts via HIF1 activation, metabolically promoting tumor growth.Cell Cycle. 2012 Jun 15;11(12):2272-84. doi: 10.4161/cc.20717. Epub 2012 Jun 15. Cell Cycle. 2012. PMID: 22684333 Free PMC article.
-
Pyruvate kinase expression (PKM1 and PKM2) in cancer-associated fibroblasts drives stromal nutrient production and tumor growth.Cancer Biol Ther. 2011 Dec 15;12(12):1101-13. doi: 10.4161/cbt.12.12.18703. Epub 2011 Dec 15. Cancer Biol Ther. 2011. PMID: 22236875 Free PMC article.
-
Mitochondrial oxidative stress in cancer-associated fibroblasts drives lactate production, promoting breast cancer tumor growth: understanding the aging and cancer connection.Cell Cycle. 2011 Dec 1;10(23):4065-73. doi: 10.4161/cc.10.23.18254. Epub 2011 Dec 1. Cell Cycle. 2011. PMID: 22129993 Free PMC article.
-
Tumor microenvironment and metabolic synergy in breast cancers: critical importance of mitochondrial fuels and function.Semin Oncol. 2014 Apr;41(2):195-216. doi: 10.1053/j.seminoncol.2014.03.002. Epub 2014 Mar 5. Semin Oncol. 2014. PMID: 24787293 Review.
-
Stromal-epithelial metabolic coupling in cancer: integrating autophagy and metabolism in the tumor microenvironment.Int J Biochem Cell Biol. 2011 Jul;43(7):1045-51. doi: 10.1016/j.biocel.2011.01.023. Epub 2011 Feb 15. Int J Biochem Cell Biol. 2011. PMID: 21300172 Free PMC article. Review.
Cited by
-
Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer.Signal Transduct Target Ther. 2021 Jun 10;6(1):218. doi: 10.1038/s41392-021-00641-0. Signal Transduct Target Ther. 2021. PMID: 34108441 Free PMC article. Review.
-
Metabolic Relationship Between Cancer-Associated Fibroblasts and Cancer Cells.Adv Exp Med Biol. 2021;1311:189-204. doi: 10.1007/978-3-030-65768-0_14. Adv Exp Med Biol. 2021. PMID: 34014544 Free PMC article.
-
Innate immune checkpoint inhibitor resistance is associated with melanoma sub-types exhibiting invasive and de-differentiated gene expression signatures.Front Immunol. 2022 Sep 28;13:955063. doi: 10.3389/fimmu.2022.955063. eCollection 2022. Front Immunol. 2022. PMID: 36248850 Free PMC article.
-
BRCA1 mutations drive oxidative stress and glycolysis in the tumor microenvironment: implications for breast cancer prevention with antioxidant therapies.Cell Cycle. 2012 Dec 1;11(23):4402-13. doi: 10.4161/cc.22776. Epub 2012 Nov 21. Cell Cycle. 2012. PMID: 23172369 Free PMC article.
-
The Therapeutic Mechanism of Schisandrol A and Its Metabolites on Pulmonary Fibrosis Based on Plasma Metabonomics and Network Analysis.Drug Des Devel Ther. 2023 Feb 15;17:477-496. doi: 10.2147/DDDT.S391503. eCollection 2023. Drug Des Devel Ther. 2023. PMID: 36814892 Free PMC article.
References
-
- Rønnov-Jessen L, Petersen OW, Bissell MJ. Cellular changes involved in conversion of normal to malignant breast: importance of the stromal reaction. Physiol Rev. 1996;76:69–125. - PubMed
-
- Rønnov-Jessen L, Petersen OW. Induction of alpha-smooth muscle actin by transforming growth factor-beta 1 in quiescent human breast gland fibroblasts. Implications for myofibroblast generation in breast neoplasia. Lab Invest. 1993;68:696–707. - PubMed
-
- Löhr M, Schmidt C, Ringel J, Kluth M, Müller P, Nizze H, et al. Transforming growth factor-beta1 induces desmoplasia in an experimental model of human pancreatic carcinoma. Cancer Res. 2001;61:550–5. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 CA075503/CA/NCI NIH HHS/United States
- R01 CA098779/CA/NCI NIH HHS/United States
- R01-CA-120876/CA/NCI NIH HHS/United States
- R01 CA120876/CA/NCI NIH HHS/United States
- R01-CA-70896/CA/NCI NIH HHS/United States
- R01-CA-098779/CA/NCI NIH HHS/United States
- R01-CA-86072/CA/NCI NIH HHS/United States
- R01-AR-055660/AR/NIAMS NIH HHS/United States
- R01-CA-080250/CA/NCI NIH HHS/United States
- R01 CA070896/CA/NCI NIH HHS/United States
- R01 CA107382/CA/NCI NIH HHS/United States
- P30 CA056036/CA/NCI NIH HHS/United States
- P30-CA-56036/CA/NCI NIH HHS/United States
- R01-CA-107382/CA/NCI NIH HHS/United States
- R01 AR055660/AR/NIAMS NIH HHS/United States
- R01-CA-75503/CA/NCI NIH HHS/United States
- R01 CA080250/CA/NCI NIH HHS/United States
- R01 CA086072/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Research Materials