

Anti-NMDA receptor encephalitis antibody binding is dependent on amino acid identity of a small region within the GluN1 amino terminal domain
- PMID: 22875940
- PMCID: PMC3430387
- DOI: 10.1523/JNEUROSCI.0064-12.2012
Anti-NMDA receptor encephalitis antibody binding is dependent on amino acid identity of a small region within the GluN1 amino terminal domain
Abstract
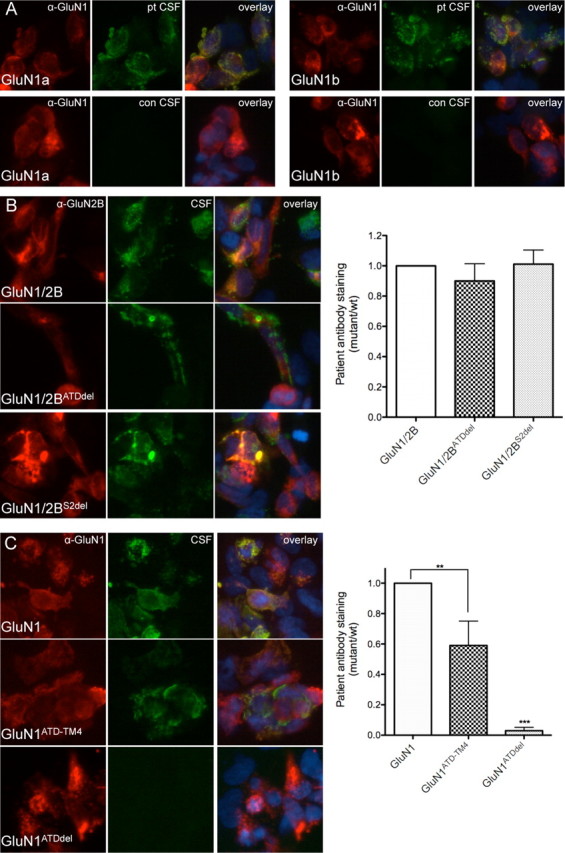
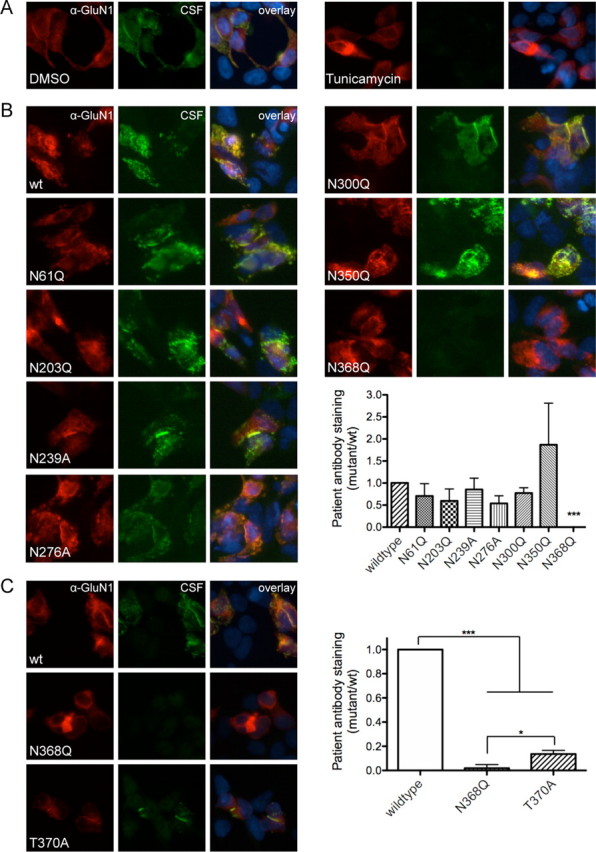
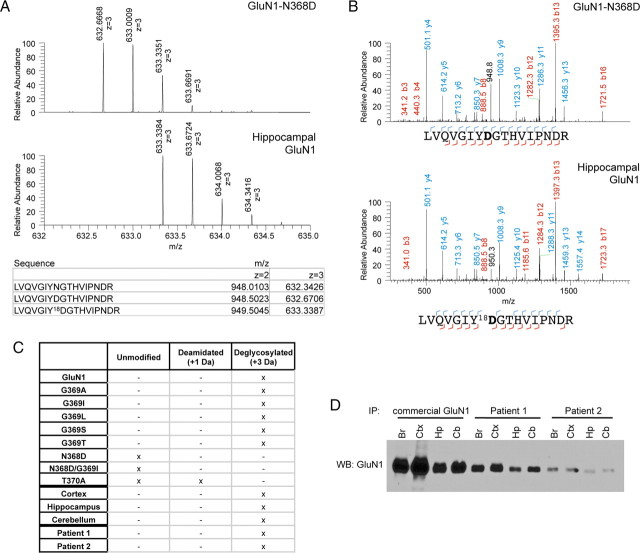
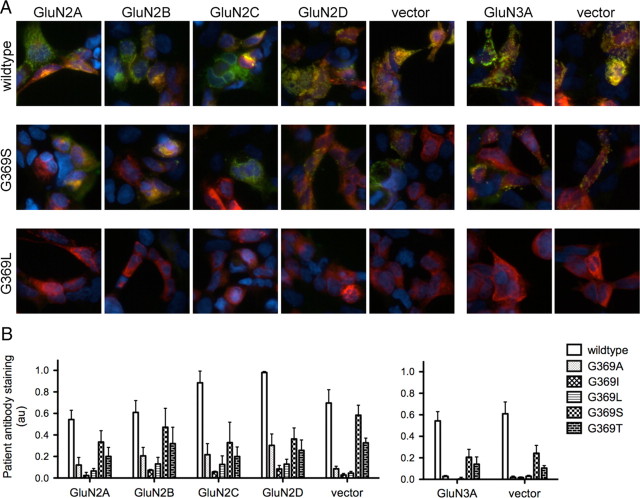
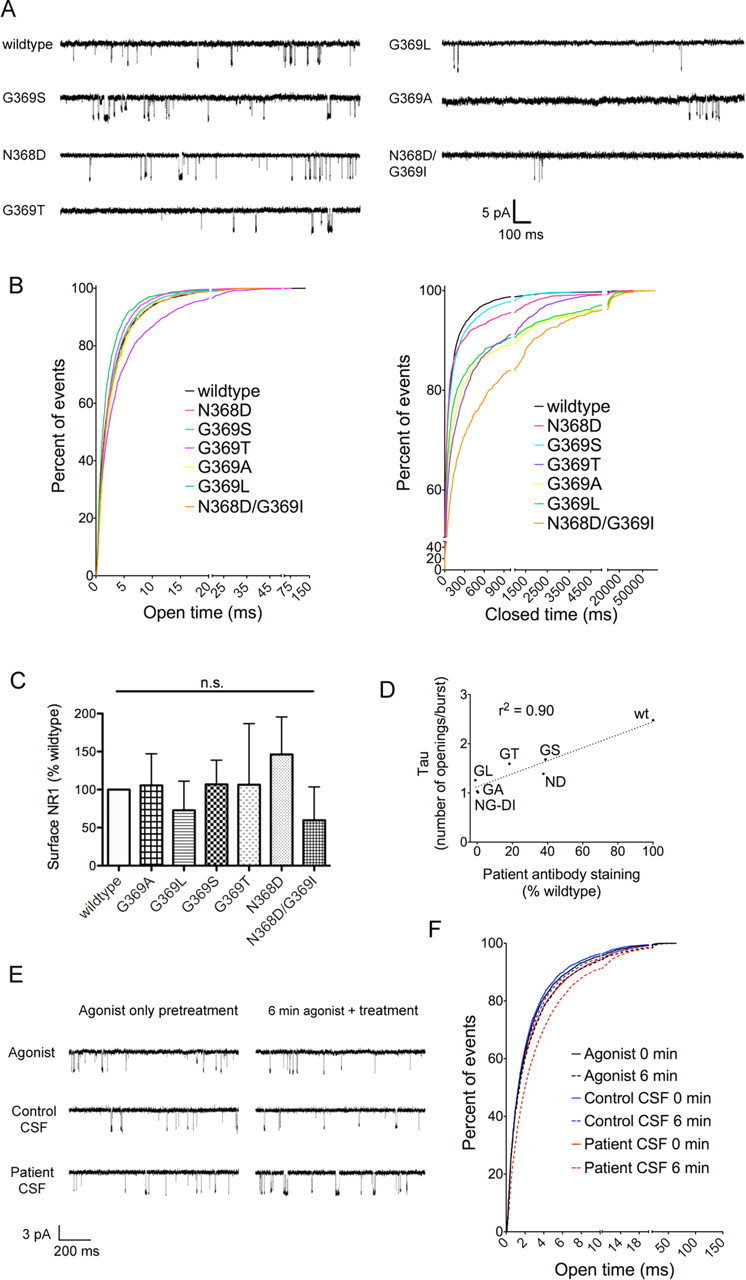
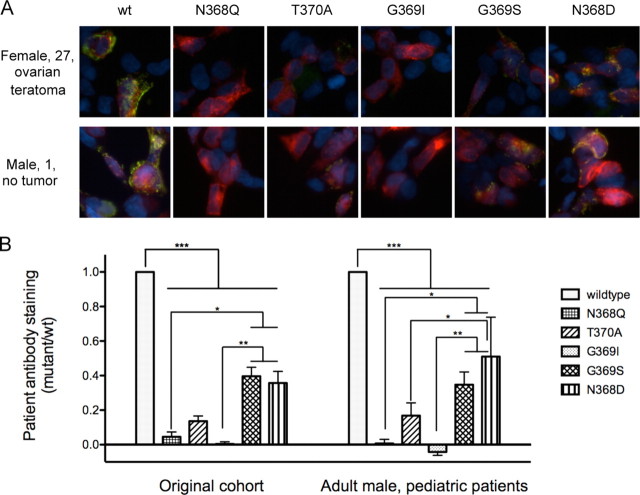
Anti-NMDA receptor (NMDAR) encephalitis is a newly identified autoimmune disorder that targets NMDARs, causing severe neurological symptoms including hallucinations, psychosis, and seizures, and may result in death (Dalmau et al., 2008). However, the exact epitope to which these antibodies bind is unknown. A clearly defined antigenic region could provide more precise testing, allow for comparison of immunogenicity between patients to explore potential clinically relevant variations, elucidate the functional effects of antibodies, and make patients' antibodies a more effective tool with which to study NMDAR function. Here, we use human CSF to explore the antigenic region of the NMDAR. We created a series of mutants within the amino terminal domain of GluN1 that change patient antibody binding in transfected cells in stereotyped ways. These mutants demonstrate that the N368/G369 region of GluN1 is crucial for the creation of immunoreactivity. Mass spectrometry experiments show that N368 is glycosylated in transfected cells and rat brain regions; however, this glycosylation is not directly required for epitope formation. Mutations of residues N368/G369 change the closed time of the receptor in single channel recordings; more frequent channel openings correlates with the degree of antibody staining, and acute antibody exposure prolongs open time of the receptor. The staining pattern of mutant receptors is similar across subgroups of patients, indicating consistent immunogenicity, although we have identified one region that has a variable role in epitope formation. These findings provide tools for detailed comparison of antibodies across patients and suggest an interaction between antibody binding and channel function.
Figures


References
-
- Akazawa C, Shigemoto R, Bessho Y, Nakanishi S, Mizuno N. Differential expression of five N-methyl-d-aspartate receptor subunit mRNAs in the cerebellum of developing and adult rats. J Comp Neurol. 1994;347:150–160. - PubMed
-
- Chapman MR, Vause HE. Anti-NMDA receptor encephalitis: diagnosis, psychiatric presentation, and treatment. Am J Psychiatry. 2011;168:245–251. - PubMed
-
- Chazot PL, Cik M, Stephenson FA. An investigation into the role of N-glycosylation in the functional expression of a recombinant heteromeric NMDA receptor. Mol Membr Biol. 1995;12:331–337. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R21 NS081439/NS/NINDS NIH HHS/United States
- NS067270/NS/NINDS NIH HHS/United States
- R56 CA089054/CA/NCI NIH HHS/United States
- P30 HD026979/HD/NICHD NIH HHS/United States
- F31 MH083395/MH/NIMH NIH HHS/United States
- CA89054/CA/NCI NIH HHS/United States
- R01 MH094741/MH/NIMH NIH HHS/United States
- R01 NS077851/NS/NINDS NIH HHS/United States
- R01 NS045986/NS/NINDS NIH HHS/United States
- R21 NS067270/NS/NINDS NIH HHS/United States
- R01 CA089054/CA/NCI NIH HHS/United States
- MH083395/MH/NIMH NIH HHS/United States
- NS077851/NS/NINDS NIH HHS/United States
- NS045986/NS/NINDS NIH HHS/United States
- MH094741/MH/NIMH NIH HHS/United States
- R56 NS045986/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases