

Relating the disease mutation spectrum to the evolution of the cystic fibrosis transmembrane conductance regulator (CFTR)
- PMID: 22879944
- PMCID: PMC3413703
- DOI: 10.1371/journal.pone.0042336
Relating the disease mutation spectrum to the evolution of the cystic fibrosis transmembrane conductance regulator (CFTR)
Abstract
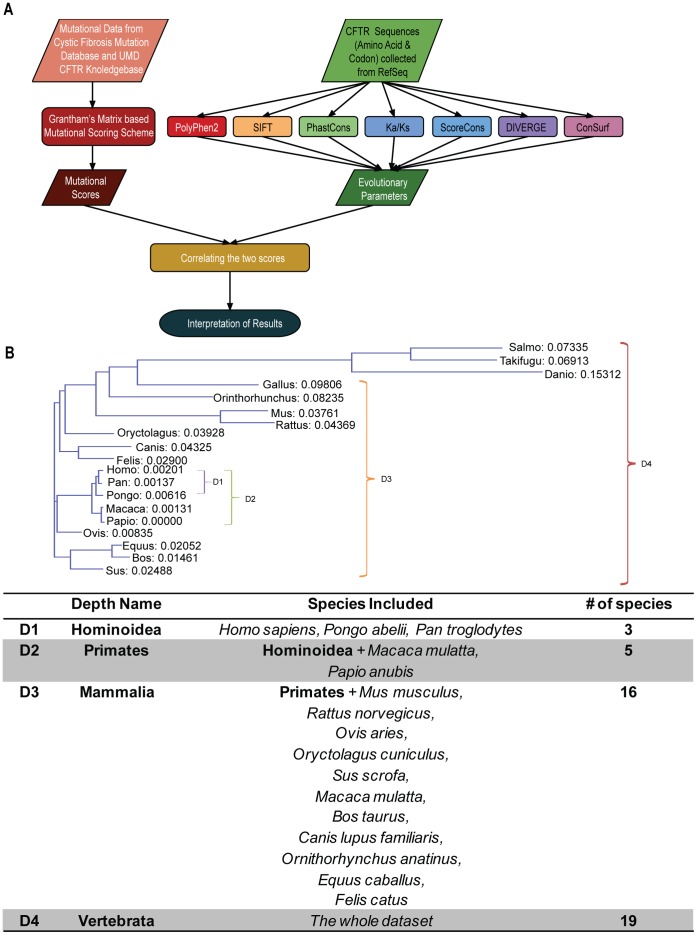
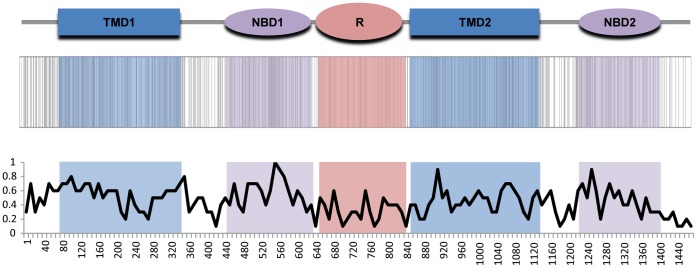
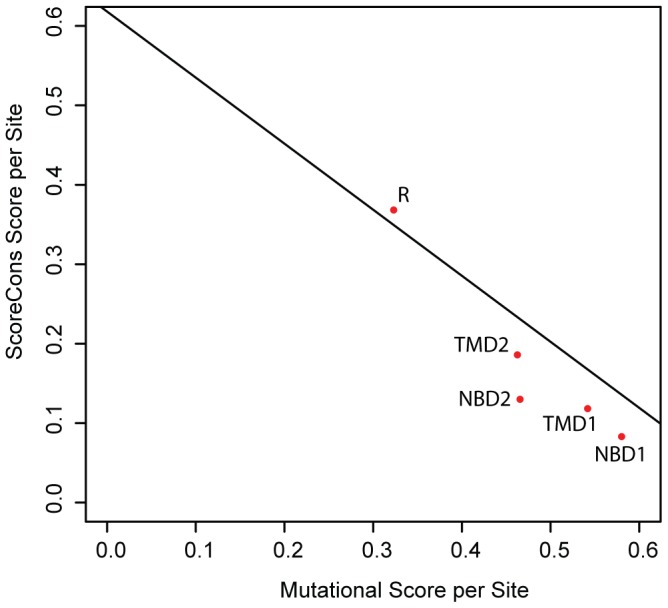
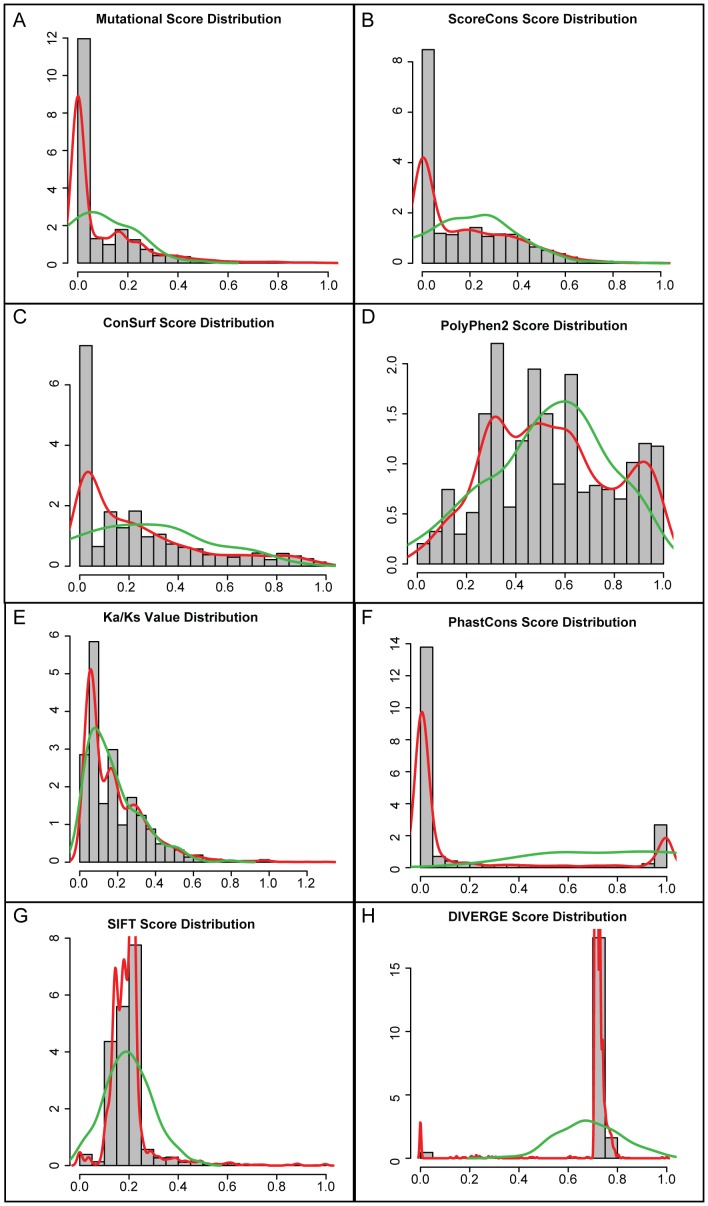
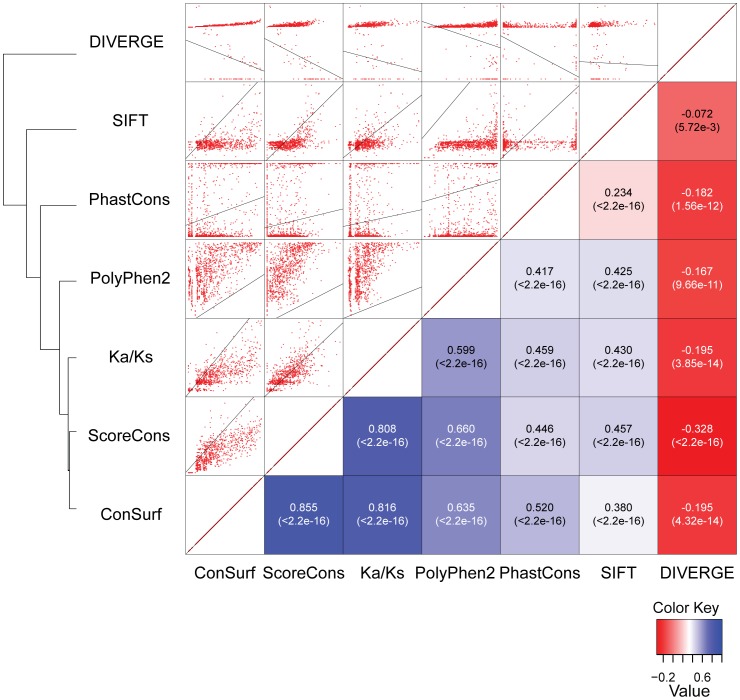
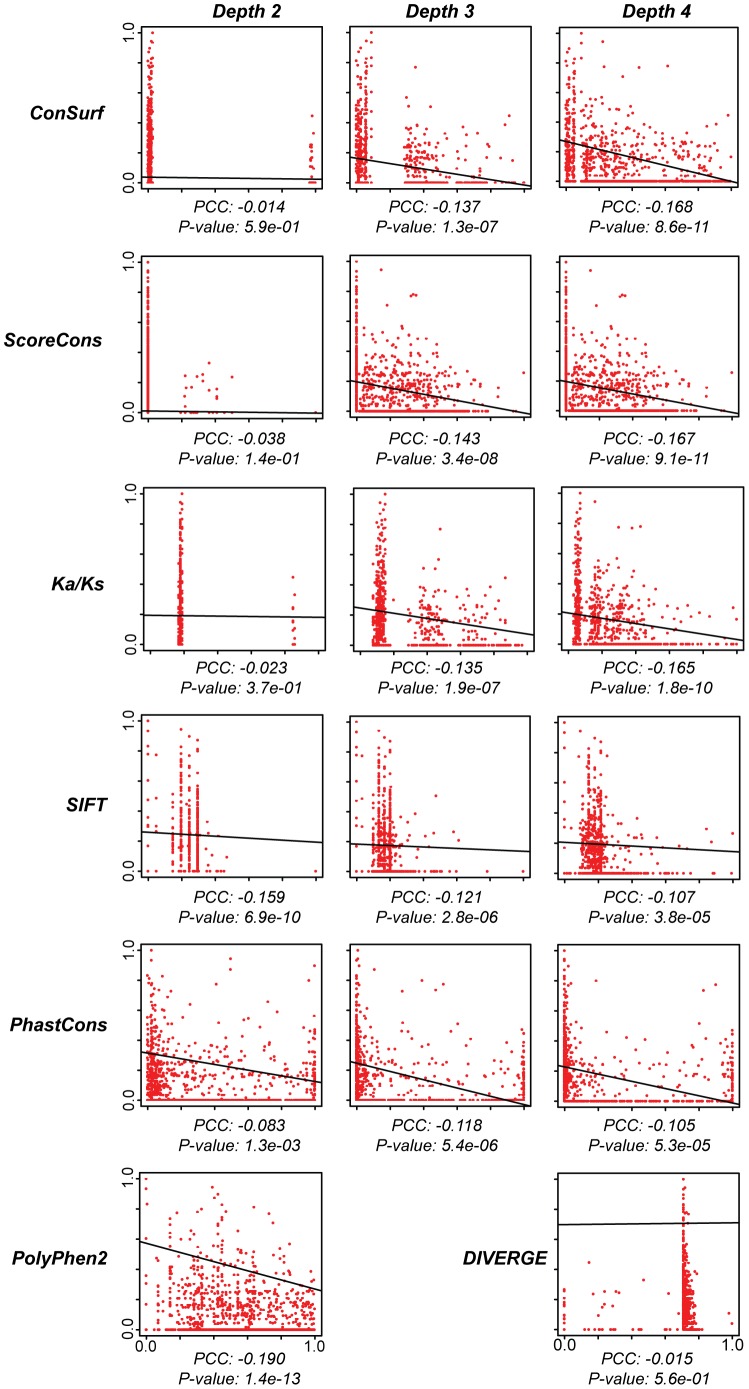
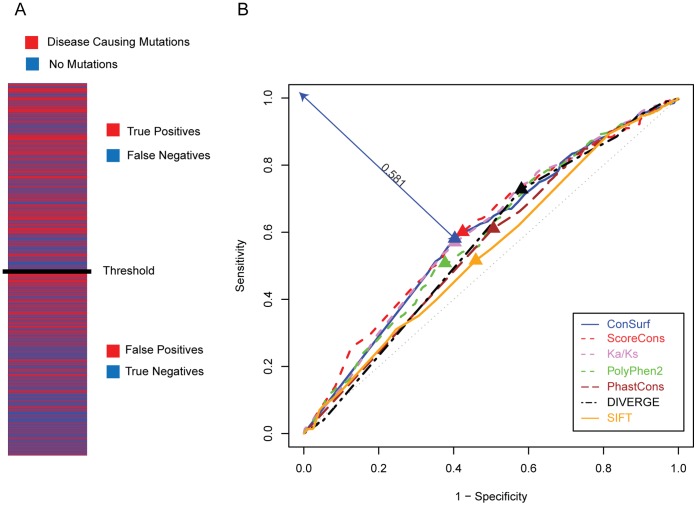
Cystic fibrosis (CF) is the most common genetic disease among Caucasians, and accordingly the cystic fibrosis transmembrane conductance regulator (CFTR) protein has perhaps the best characterized disease mutation spectrum with more than 1,500 causative mutations having been identified. In this study, we took advantage of that wealth of mutational information in an effort to relate site-specific evolutionary parameters with the propensity and severity of CFTR disease-causing mutations. To do this, we devised a scoring scheme for known CFTR disease-causing mutations based on the Grantham amino acid chemical difference matrix. CFTR site-specific evolutionary constraint values were then computed for seven different evolutionary metrics across a range of increasing evolutionary depths. The CFTR mutational scores and the various site-specific evolutionary constraint values were compared in order to evaluate which evolutionary measures best reflect the disease-causing mutation spectrum. Site-specific evolutionary constraint values from the widely used comparative method PolyPhen2 show the best correlation with the CFTR mutation score spectrum, whereas more straightforward conservation based measures (ConSurf and ScoreCons) show the greatest ability to predict individual CFTR disease-causing mutations. While far greater than could be expected by chance alone, the fraction of the variability in mutation scores explained by the PolyPhen2 metric (3.6%), along with the best set of paired sensitivity (58%) and specificity (60%) values for the prediction of disease-causing residues, were marginal. These data indicate that evolutionary constraint levels are informative but far from determinant with respect to disease-causing mutations in CFTR. Nevertheless, this work shows that, when combined with additional lines of evidence, information on site-specific evolutionary conservation can and should be used to guide site-directed mutagenesis experiments by more narrowly defining the set of target residues, resulting in a potential savings of both time and money.
Conflict of interest statement
Figures
References
-
- Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, et al. (1989) Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245: 1066–1073. - PubMed
-
- Zieve D, Hadjiliadis D (2011) Cystic Fibrosis. Available: http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0001167/. Accessed 2012 Mar 30..
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
