

IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress
- PMID: 22883233
- PMCID: PMC4014071
- DOI: 10.1016/j.cmet.2012.07.007
IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress
Abstract
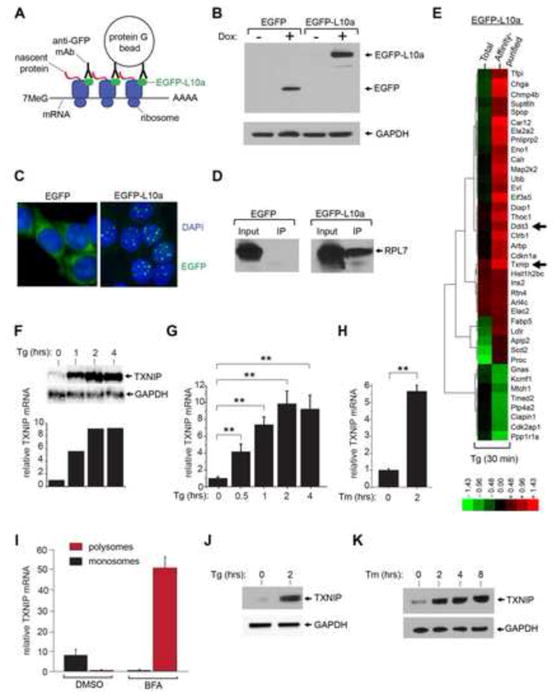
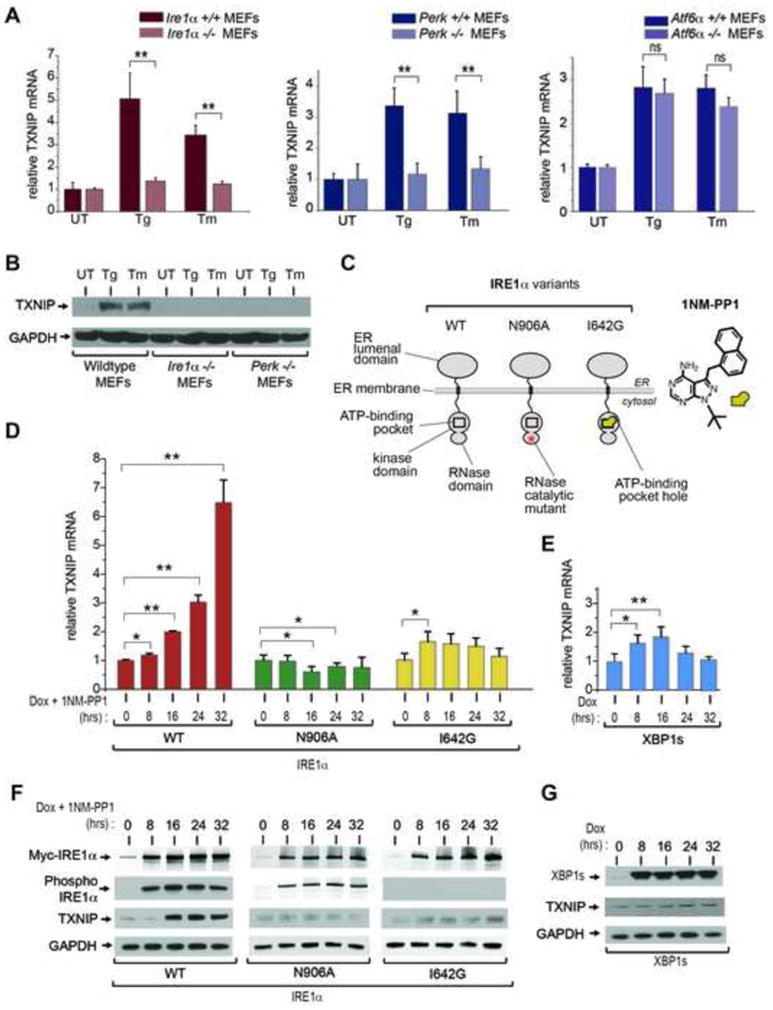
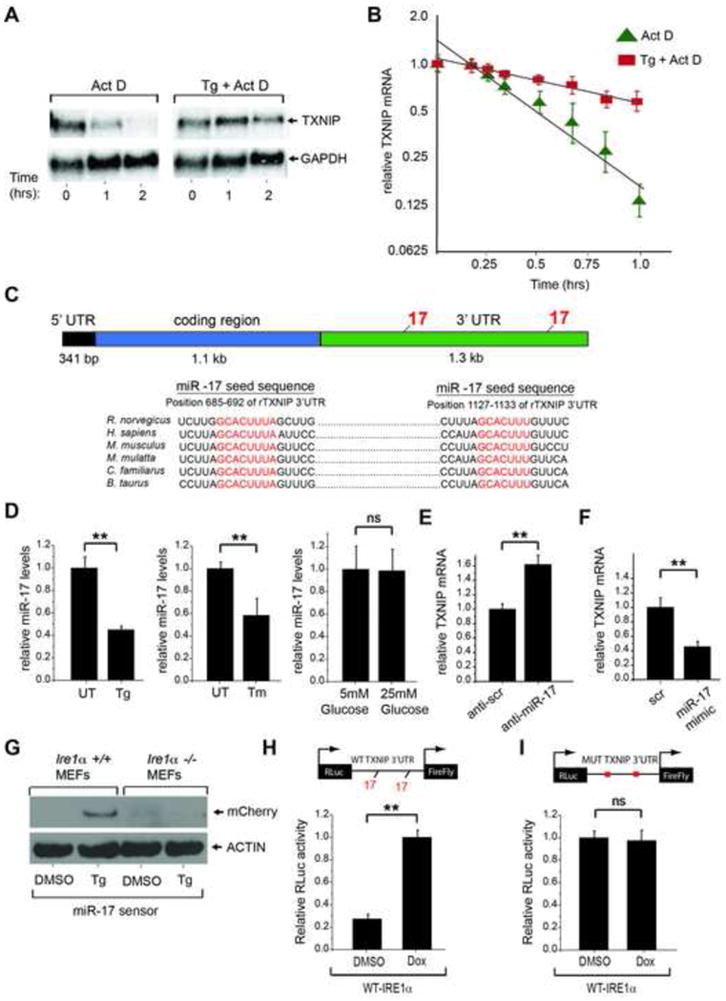
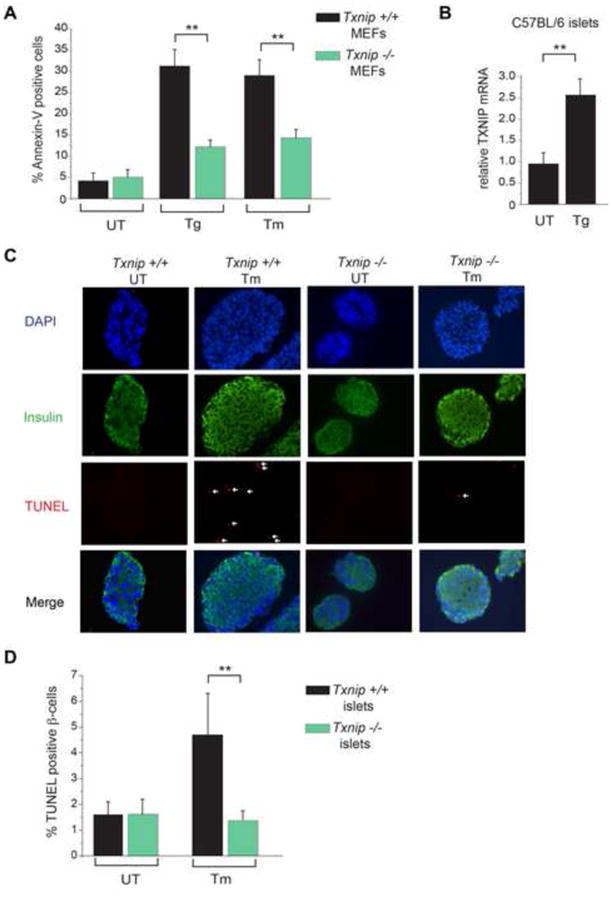
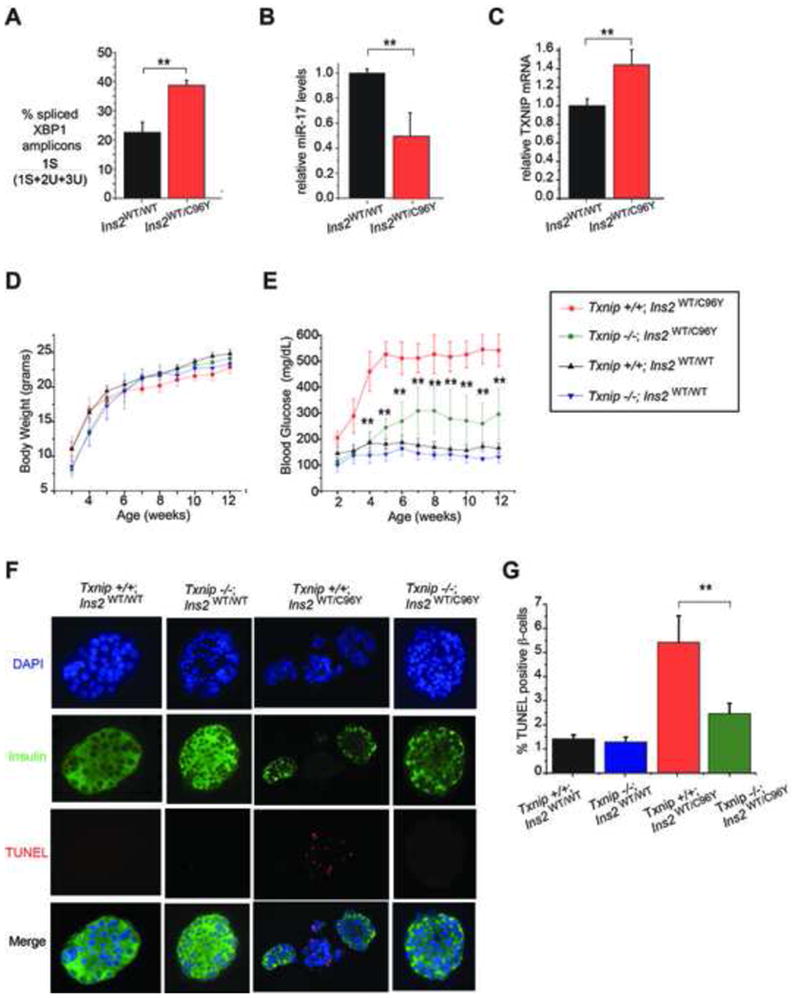
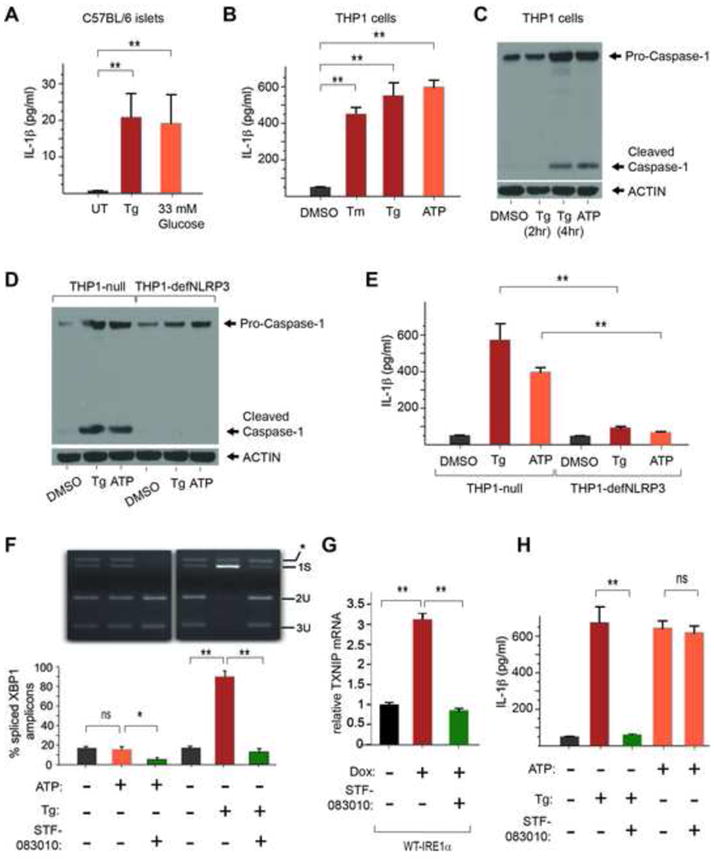
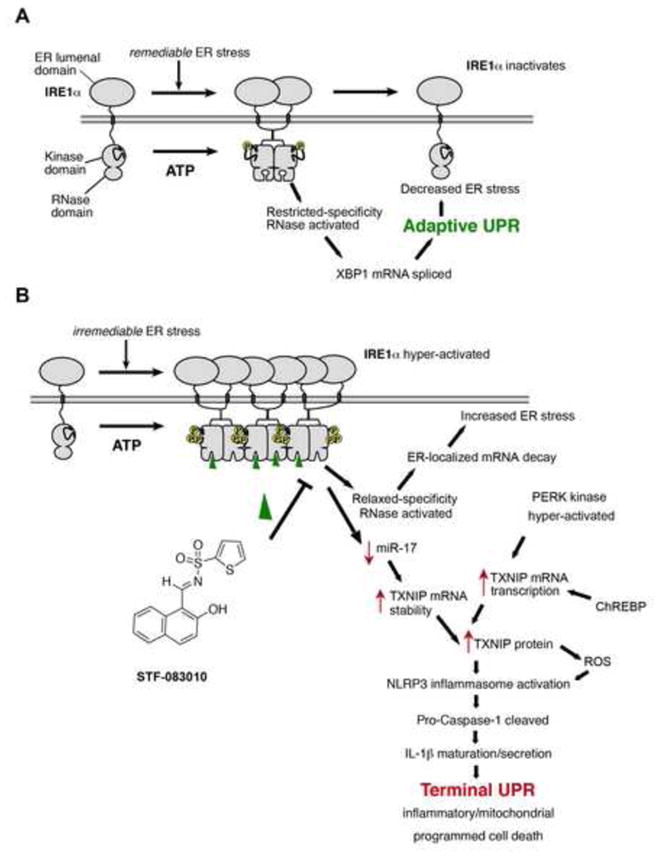
When unfolded proteins accumulate to irremediably high levels within the endoplasmic reticulum (ER), intracellular signaling pathways called the unfolded protein response (UPR) become hyperactivated to cause programmed cell death. We discovered that thioredoxin-interacting protein (TXNIP) is a critical node in this "terminal UPR." TXNIP becomes rapidly induced by IRE1α, an ER bifunctional kinase/endoribonuclease (RNase). Hyperactivated IRE1α increases TXNIP mRNA stability by reducing levels of a TXNIP destabilizing microRNA, miR-17. In turn, elevated TXNIP protein activates the NLRP3 inflammasome, causing procaspase-1 cleavage and interleukin 1β (IL-1β) secretion. Txnip gene deletion reduces pancreatic β cell death during ER stress and suppresses diabetes caused by proinsulin misfolding in the Akita mouse. Finally, small molecule IRE1α RNase inhibitors suppress TXNIP production to block IL-1β secretion. In summary, the IRE1α-TXNIP pathway is used in the terminal UPR to promote sterile inflammation and programmed cell death and may be targeted to develop effective treatments for cell degenerative diseases.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures
Comment in
-
TXNIP switches tracks toward a terminal UPR.Cell Metab. 2012 Aug 8;16(2):135-7. doi: 10.1016/j.cmet.2012.07.012. Cell Metab. 2012. PMID: 22883225
References
-
- Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
