

Diabetic peripheral neuropathy: should a chaperone accompany our therapeutic approach?
- PMID: 22885705
- PMCID: PMC3462992
- DOI: 10.1124/pr.111.005314
Diabetic peripheral neuropathy: should a chaperone accompany our therapeutic approach?
Abstract
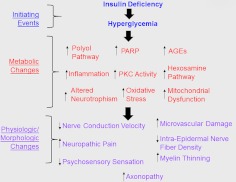
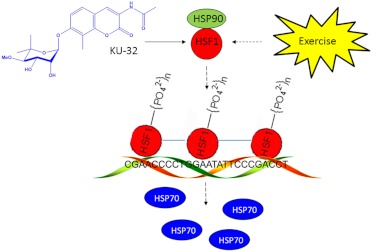
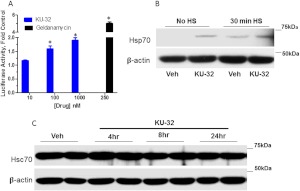
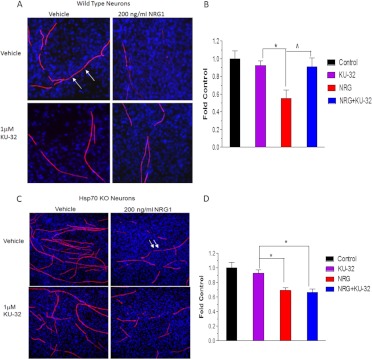
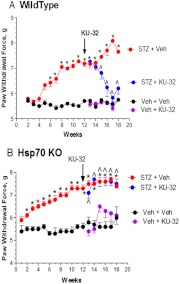
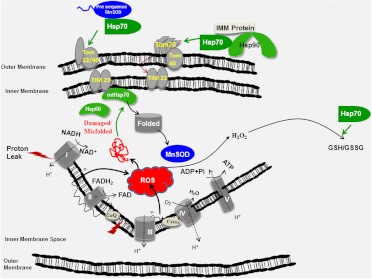
Diabetic peripheral neuropathy (DPN) is a common complication of diabetes that is associated with axonal atrophy, demyelination, blunted regenerative potential, and loss of peripheral nerve fibers. The development and progression of DPN is due in large part to hyperglycemia but is also affected by insulin deficiency and dyslipidemia. Although numerous biochemical mechanisms contribute to DPN, increased oxidative/nitrosative stress and mitochondrial dysfunction seem intimately associated with nerve dysfunction and diminished regenerative capacity. Despite advances in understanding the etiology of DPN, few approved therapies exist for the pharmacological management of painful or insensate DPN. Therefore, identifying novel therapeutic strategies remains paramount. Because DPN does not develop with either temporal or biochemical uniformity, its therapeutic management may benefit from a multifaceted approach that inhibits pathogenic mechanisms, manages inflammation, and increases cytoprotective responses. Finally, exercise has long been recognized as a part of the therapeutic management of diabetes, and exercise can delay and/or prevent the development of painful DPN. This review presents an overview of existing therapies that target both causal and symptomatic features of DPN and discusses the role of up-regulating cytoprotective pathways via modulating molecular chaperones. Overall, it may be unrealistic to expect that a single pharmacologic entity will suffice to ameliorate the multiple symptoms of human DPN. Thus, combinatorial therapies that target causal mechanisms and enhance endogenous reparative capacity may enhance nerve function and improve regeneration in DPN if they converge to decrease oxidative stress, improve mitochondrial bioenergetics, and increase response to trophic factors.
Figures
References
-
- Abbas ZG, Swai AB. (1997) Evaluation of the efficacy of thiamine and pyridoxine in the treatment of symptomatic diabetic peripheral neuropathy. East Afr Med J 74:803–808 - PubMed
-
- PKC-DRS2 Group, Aiello LP, Davis MD, Girach A, Kles KA, Milton RC, Sheetz MJ, Vignati L, Zhi XE. (2006) Effect of ruboxistaurin on visual loss in patients with diabetic retinopathy. Ophthalmology 113:2221–2230 - PubMed
-
- Akkina SK, Patterson CL, Wright DE. (2001) GDNF rescues nonpeptidergic unmyelinated primary afferents in streptozotocin-treated diabetic mice. Exp Neurol 167:173–182 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
