

Pathogenic mechanisms of acute pancreatitis
- PMID: 22885948
- PMCID: PMC3682674
- DOI: 10.1097/MOG.0b013e3283567f52
Pathogenic mechanisms of acute pancreatitis
Abstract
Purpose of review: In this article, recent advances in the pathogenesis of acute pancreatitis have been reviewed.
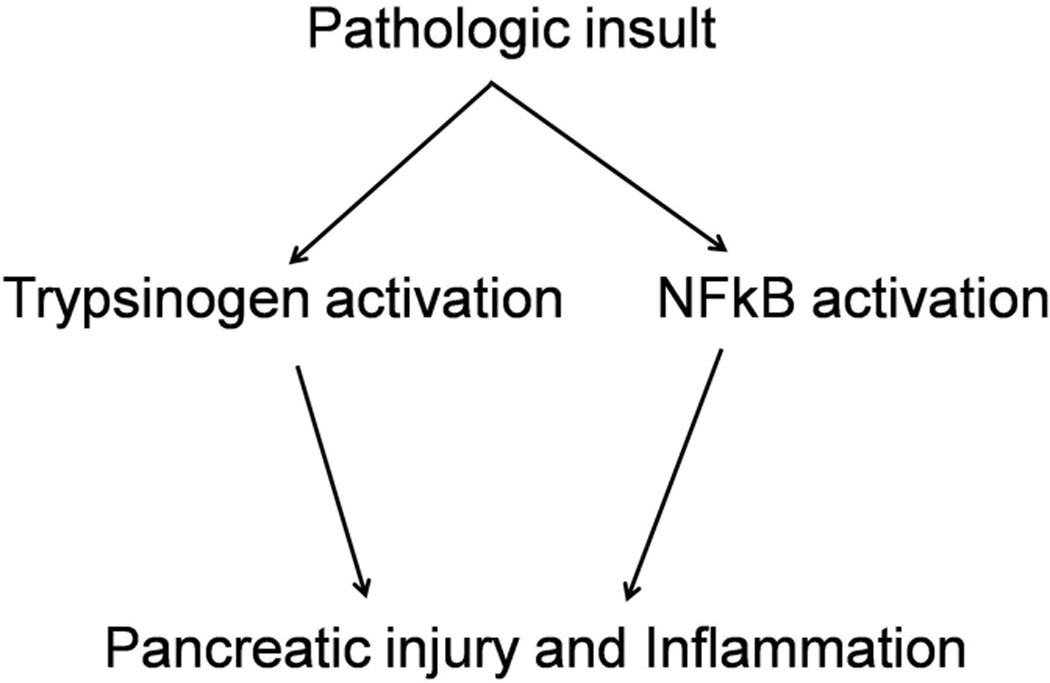
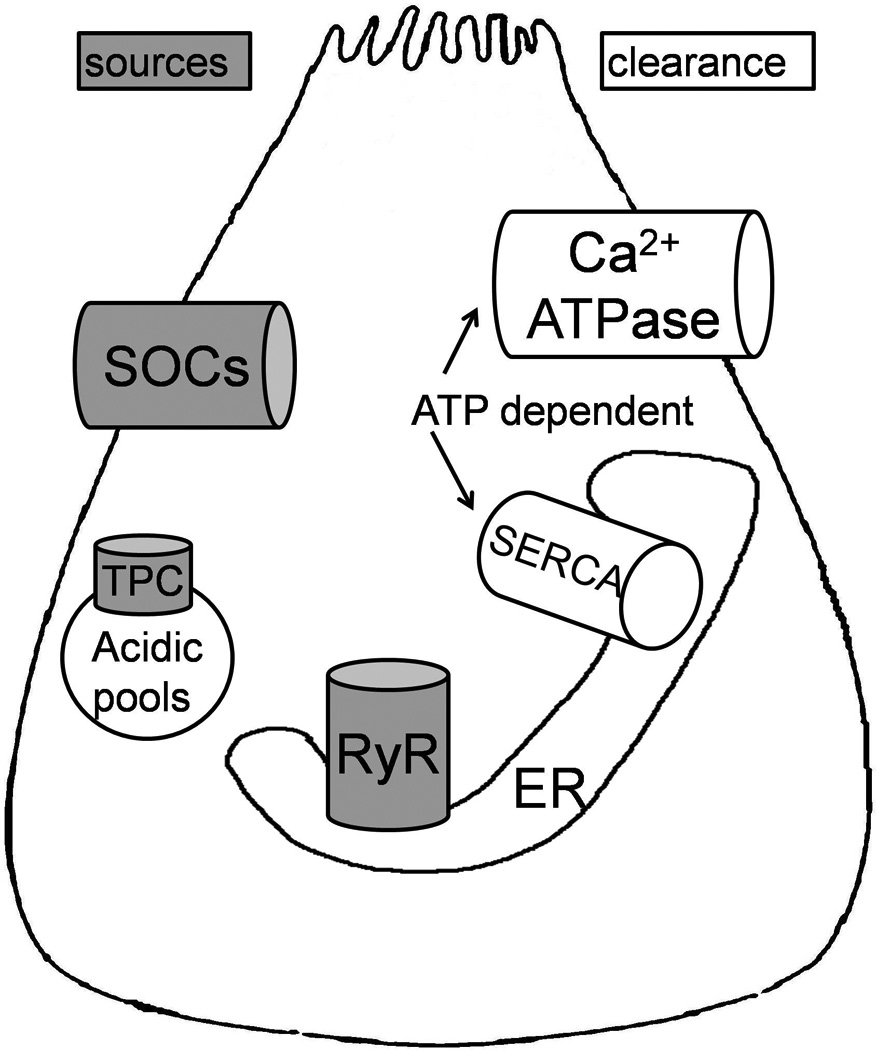
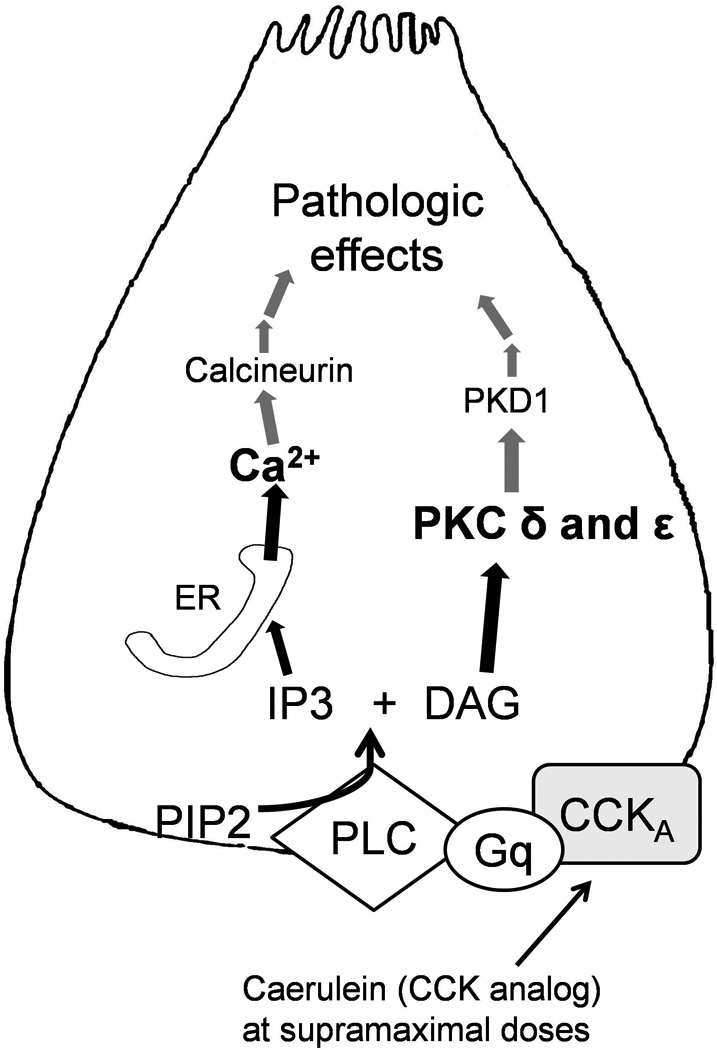
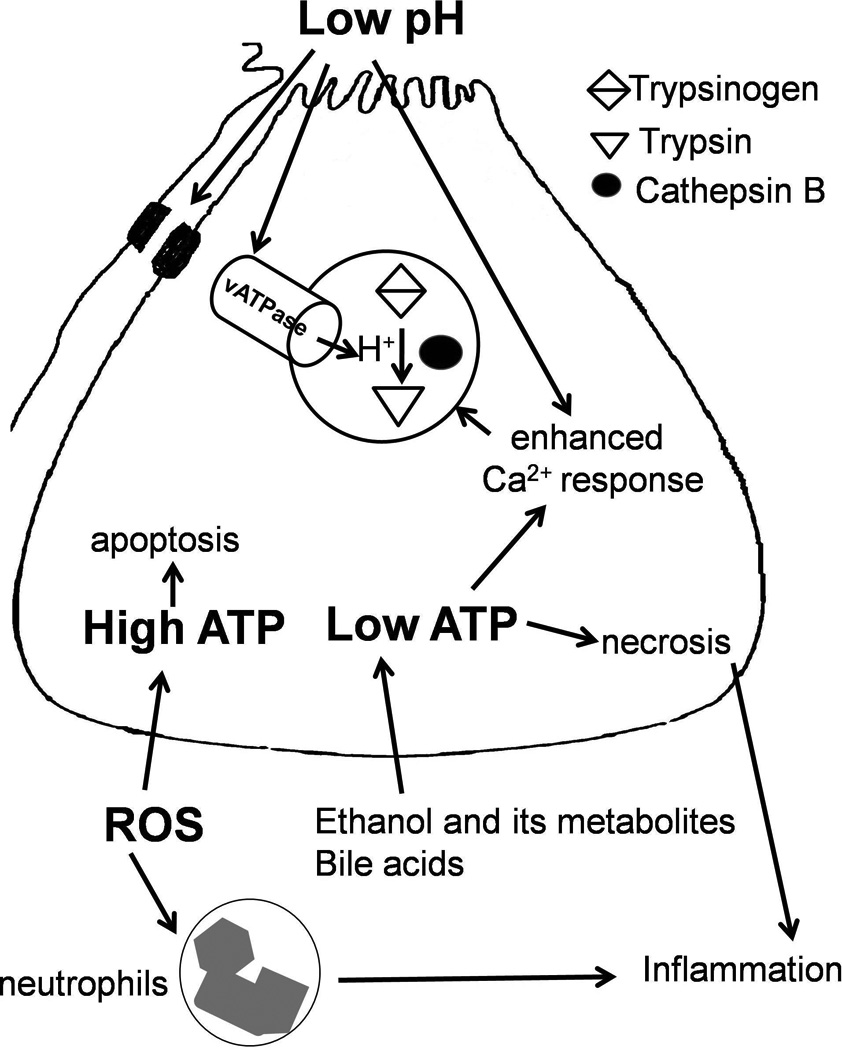
Recent findings: Pathologic intra-acinar trypsinogen activation had been hypothesized to be the central mechanism of pancreatitis for over a century. This hypothesis could be explored for the first time with the development of a novel mouse model lacking pathologic intra-acinar trypsinogen activation. It became clear that intra-acinar trypsinogen activation contributes to early acinar injury, but local and systemic inflammation progress independently during pancreatitis. Early intra-acinar nuclear factor kappa B (NFκB) activation, which occurs parallel to but independent of trypsinogen activation, may be crucial in pancreatitis. Although the mechanism of NFκB and trypsinogen activation is not entirely clear, further insights have been made into key pathogenic cellular events such as calcium signaling, mitochondrial dysfunction, endoplasmic reticulum (ER) stress, autophagy and impaired trafficking, and lysosomal and secretory responses. Cellular intrinsic damage-sensing mechanisms that lead to activation of the inflammatory response aimed at repair, but lead to disease when overwhelmed, are beginning to be understood.
Summary: New findings necessitate a paradigm shift in our understanding of acute pancreatitis. Intra-acinar trypsinogen activation leads to early pancreatic injury, but the inflammatory response of acute pancreatitis develops independently, driven by early activation of inflammatory pathways.
Conflict of interest statement
Conflict of Interest: None to disclose.
Figures
References
-
- Chiari H. ÜberdieSelbstverdauung des menschlichen Pankreas. Zeitschriftfür Heilkunde. 1896;17:69–96.
-
- Halangk W, Lerch MM. Early events in acute pancreatitis. Clin Lab Med. 2005;25(1):1–15. - PubMed
-
- Saluja AK, Lerch MM, Phillips PA, Dudeja V. Why does pancreatic overstimulation cause pancreatitis? Annu Rev Physiol. 2007;69:249–269. - PubMed
-
- Hofbauer B, Saluja AK, Lerch MM, Bhagat L, Bhatia M, Lee HS, Frossard JL, Adler G, Steer ML. Intra-acinar cell activation of trypsinogen during caerulein-induced pancreatitis in rats. Am J Physiol. 1998;275(2 Pt 1):G352–G362. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
