

Induction of cardiac fibrosis by β-blocker in G protein-independent and G protein-coupled receptor kinase 5/β-arrestin2-dependent Signaling pathways
- PMID: 22888001
- PMCID: PMC3471751
- DOI: 10.1074/jbc.M112.357871
Induction of cardiac fibrosis by β-blocker in G protein-independent and G protein-coupled receptor kinase 5/β-arrestin2-dependent Signaling pathways
Abstract
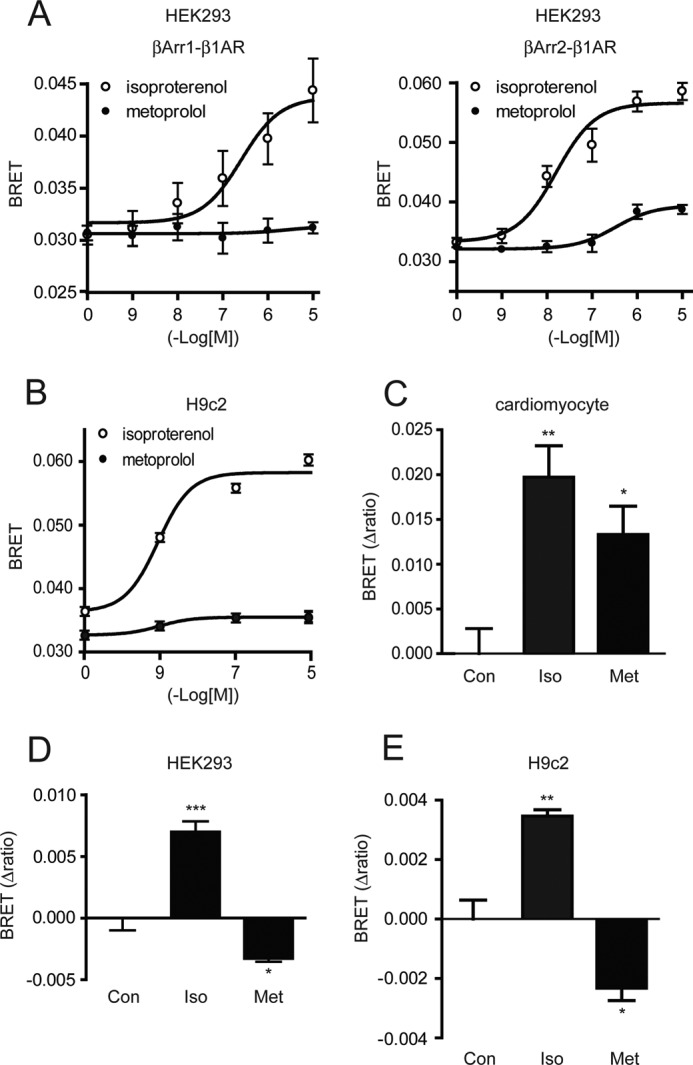
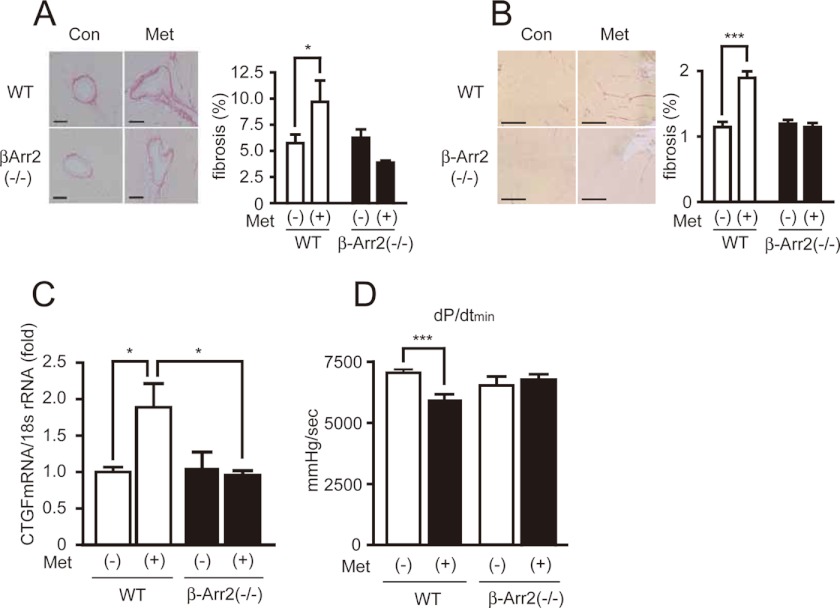
G-protein coupled receptors (GPCRs) have long been known as receptors that activate G protein-dependent cellular signaling pathways. In addition to the G protein-dependent pathways, recent reports have revealed that several ligands called "biased ligands" elicit G protein-independent and β-arrestin-dependent signaling through GPCRs (biased agonism). Several β-blockers are known as biased ligands. All β-blockers inhibit the binding of agonists to the β-adrenergic receptors. In addition to β-blocking action, some β-blockers are reported to induce cellular responses through G protein-independent and β-arrestin-dependent signaling pathways. However, the physiological significance induced by the β-arrestin-dependent pathway remains much to be clarified in vivo. Here, we demonstrate that metoprolol, a β(1)-adrenergic receptor-selective blocker, could induce cardiac fibrosis through a G protein-independent and β-arrestin2-dependent pathway. Metoprolol, a β-blocker, increased the expression of fibrotic genes responsible for cardiac fibrosis in cardiomyocytes. Furthermore, metoprolol induced the interaction between β(1)-adrenergic receptor and β-arrestin2, but not β-arrestin1. The interaction between β(1)-adrenergic receptor and β-arrestin2 by metoprolol was impaired in the G protein-coupled receptor kinase 5 (GRK5)-knockdown cells. Metoprolol-induced cardiac fibrosis led to cardiac dysfunction. However, the metoprolol-induced fibrosis and cardiac dysfunction were not evoked in β-arrestin2- or GRK5-knock-out mice. Thus, metoprolol is a biased ligand that selectively activates a G protein-independent and GRK5/β-arrestin2-dependent pathway, and induces cardiac fibrosis. This study demonstrates the physiological importance of biased agonism, and suggests that G protein-independent and β-arrestin-dependent signaling is a reason for the diversity of the effectiveness of β-blockers.
Figures





Similar articles
-
beta-arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptor.J Biol Chem. 2006 Jan 13;281(2):1261-73. doi: 10.1074/jbc.M506576200. Epub 2005 Nov 9. J Biol Chem. 2006. PMID: 16280323
-
β-arrestin1-biased β1-adrenergic receptor signaling regulates microRNA processing.Circ Res. 2014 Feb 28;114(5):833-44. doi: 10.1161/CIRCRESAHA.114.302766. Epub 2013 Dec 13. Circ Res. 2014. PMID: 24334028 Free PMC article.
-
β-Arrestin-Biased Allosteric Modulator Potentiates Carvedilol-Stimulated β Adrenergic Receptor Cardioprotection.Mol Pharmacol. 2021 Dec;100(6):568-579. doi: 10.1124/molpharm.121.000359. Epub 2021 Sep 24. Mol Pharmacol. 2021. PMID: 34561298 Free PMC article.
-
Physiologic and cardiac roles of beta-arrestins.J Mol Cell Cardiol. 2009 Mar;46(3):300-8. doi: 10.1016/j.yjmcc.2008.11.015. Epub 2008 Dec 6. J Mol Cell Cardiol. 2009. PMID: 19103204 Review.
-
G Protein-Coupled Receptor Signaling Through β-Arrestin-Dependent Mechanisms.J Cardiovasc Pharmacol. 2017 Sep;70(3):142-158. doi: 10.1097/FJC.0000000000000482. J Cardiovasc Pharmacol. 2017. PMID: 28328745 Free PMC article. Review.
Cited by
-
Therapeutic Molecular Phenotype of β-Blocker-Associated Reverse-Remodeling in Nonischemic Dilated Cardiomyopathy.Circ Cardiovasc Genet. 2015 Apr;8(2):270-83. doi: 10.1161/CIRCGENETICS.114.000767. Epub 2015 Jan 30. Circ Cardiovasc Genet. 2015. PMID: 25637602 Free PMC article. Clinical Trial.
-
The emerging roles of β-arrestins in fibrotic diseases.Acta Pharmacol Sin. 2015 Nov;36(11):1277-87. doi: 10.1038/aps.2015.74. Epub 2015 Sep 21. Acta Pharmacol Sin. 2015. PMID: 26388156 Free PMC article. Review.
-
Achievement of a target dose of bisoprolol may not be a preferred option for attenuating pressure overload-induced cardiac hypertrophy and fibrosis.Exp Ther Med. 2016 Oct;12(4):2027-2038. doi: 10.3892/etm.2016.3570. Epub 2016 Aug 4. Exp Ther Med. 2016. PMID: 27698689 Free PMC article.
-
Angiotensin-converting-enzyme inhibitor prevents skeletal muscle fibrosis in myocardial infarction mice.Skelet Muscle. 2020 Apr 25;10(1):11. doi: 10.1186/s13395-020-00230-9. Skelet Muscle. 2020. PMID: 32334642 Free PMC article.
-
Tethered and Polymer Supported Bilayer Lipid Membranes: Structure and Function.Membranes (Basel). 2016 May 30;6(2):30. doi: 10.3390/membranes6020030. Membranes (Basel). 2016. PMID: 27249006 Free PMC article. Review.
References
-
- Pierce K. L., Premont R. T., Lefkowitz R. J. (2002) Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 3, 639–650 - PubMed
-
- Reiter E., Lefkowitz R. J. (2006) GRKs and β-arrestins. Roles in receptor silencing, trafficking, and signaling. Trends Endocrinol. Metab. 17, 159–165 - PubMed
-
- DeFea K. A. (2011) β-Arrestins as regulators of signal termination and transduction. How do they determine what to scaffold? Cell. Signal. 23, 621–629 - PubMed
-
- DeWire S. M., Ahn S., Lefkowitz R. J., Shenoy S. K. (2007) β-Arrestins and cell signaling. Annu. Rev. Physiol. 69, 483–510 - PubMed
-
- Premont R. T., Gainetdinov R. R. (2007) Physiological roles of G protein-coupled receptor kinases and arrestins. Annu. Rev. Physiol. 69, 511–534 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
