

PAMPs and DAMPs: signal 0s that spur autophagy and immunity
- PMID: 22889221
- PMCID: PMC3662247
- DOI: 10.1111/j.1600-065X.2012.01146.x
PAMPs and DAMPs: signal 0s that spur autophagy and immunity
Abstract
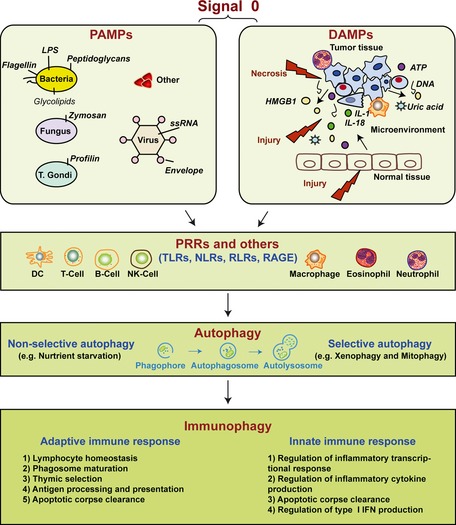
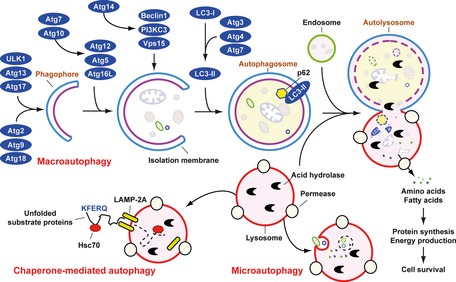
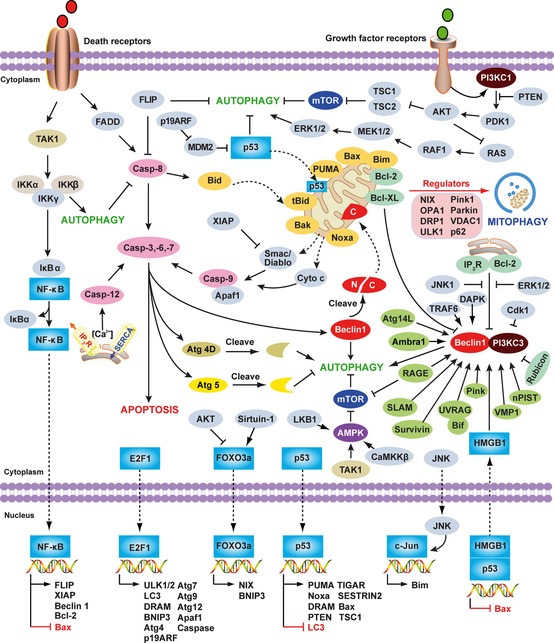
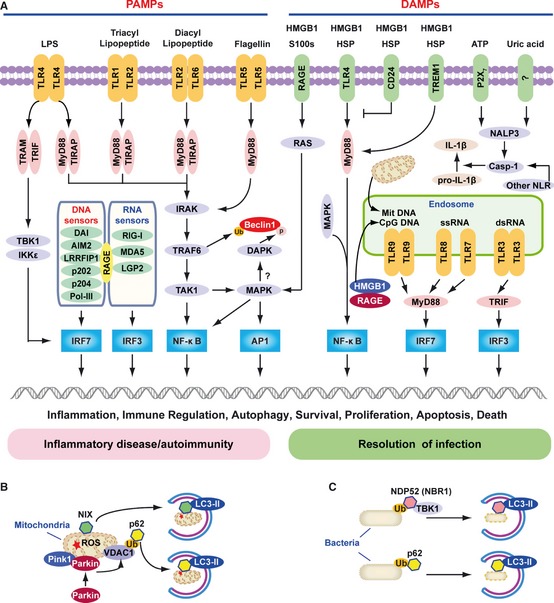
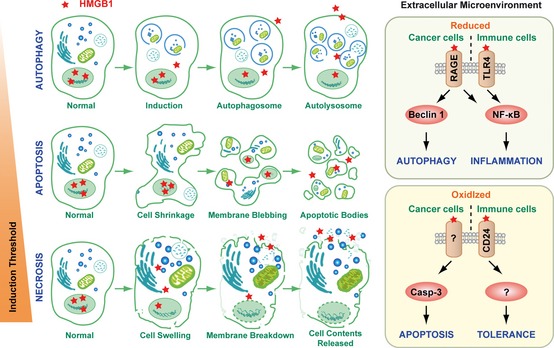
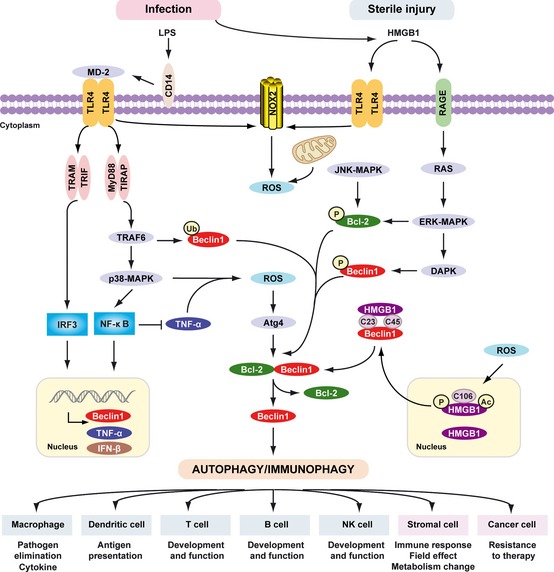
Pathogen-associated molecular pattern molecules (PAMPs) are derived from microorganisms and recognized by pattern recognition receptor (PRR)-bearing cells of the innate immune system as well as many epithelial cells. In contrast, damage-associated molecular pattern molecules (DAMPs) are cell-derived and initiate and perpetuate immunity in response to trauma, ischemia, and tissue damage, either in the absence or presence of pathogenic infection. Most PAMPs and DAMPs serve as so-called 'Signal 0s' that bind specific receptors [Toll-like receptors, NOD-like receptors, RIG-I-like receptors, AIM2-like receptors, and the receptor for advanced glycation end products (RAGE)] to promote autophagy. Autophagy, a conserved lysosomal degradation pathway, is a cell survival mechanism invoked in response to environmental and cellular stress. Autophagy is inferred to have been present in the last common eukaryotic ancestor and only to have been lost by some obligatory intracellular parasites. As such, autophagy represents a unifying biology, subserving survival and the earliest host defense strategies, predating apoptosis, within eukaryotes. Here, we review recent advances in our understanding of autophagic molecular mechanisms and functions in emergent immunity.
© 2012 John Wiley & Sons A/S.
Figures
References
-
- Janeway CA Jr. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol 1989;54:1–13. - PubMed
-
- Janeway C. Immunogenicity signals 1,2,3… and 0. Immunol Today 1989;10:283–286. - PubMed
-
- Akira S, Hemmi H. Recognition of pathogen‐associated molecular patterns by TLR family. Immunol Lett 2003;85:85–95. - PubMed
-
- Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol 1994;12:991–1045. - PubMed
-
- Rubartelli A, Lotze MT. Inside, outside, upside down: damage‐associated molecular‐pattern molecules (DAMPs) and redox. Trends Immunol 2007;28:429–436. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
