

A new conceptual paradigm for the haemodynamics of salt-sensitive hypertension: a mathematical modelling approach
- PMID: 22890716
- PMCID: PMC3530111
- DOI: 10.1113/jphysiol.2012.228619
A new conceptual paradigm for the haemodynamics of salt-sensitive hypertension: a mathematical modelling approach
Abstract
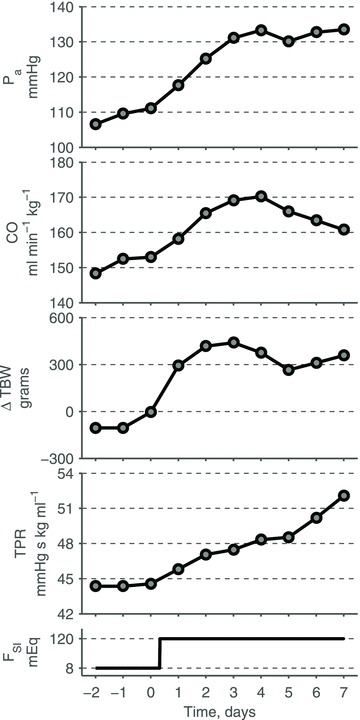
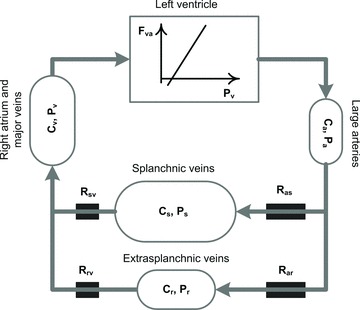
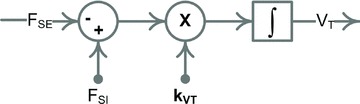
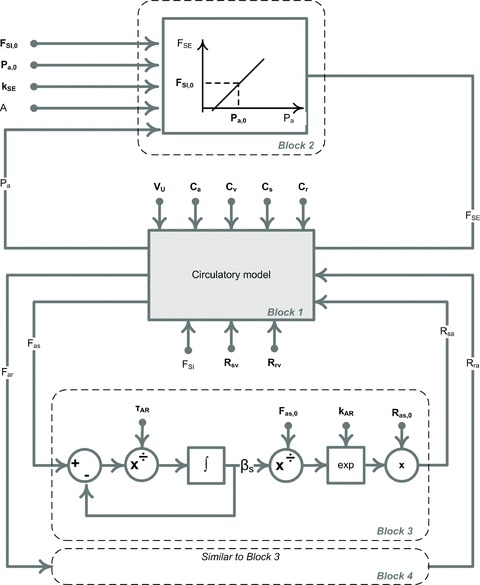
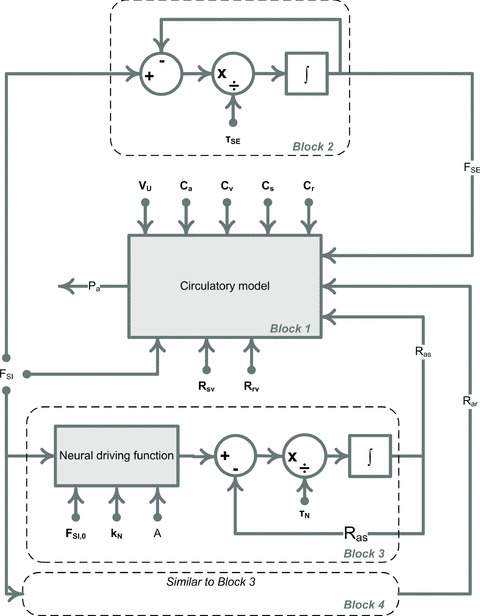
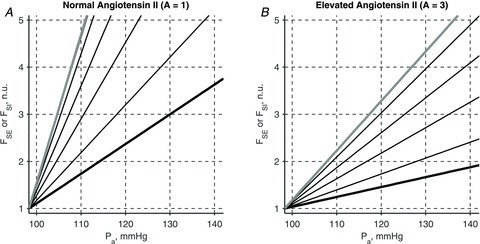
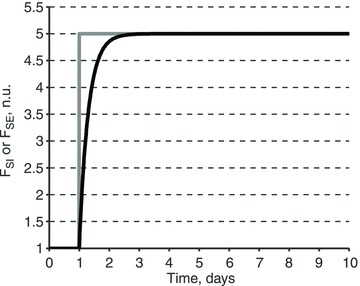
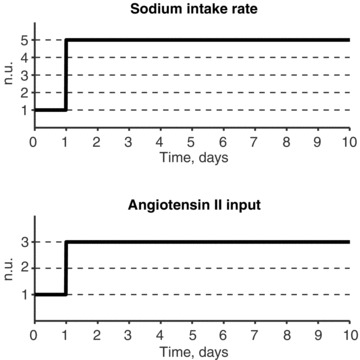
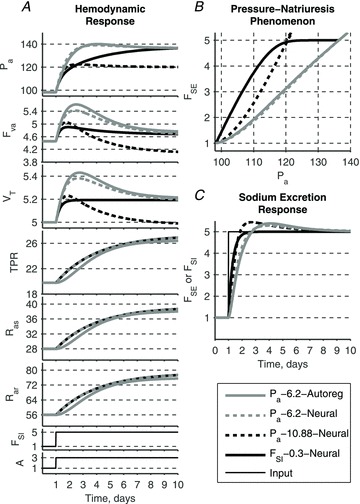
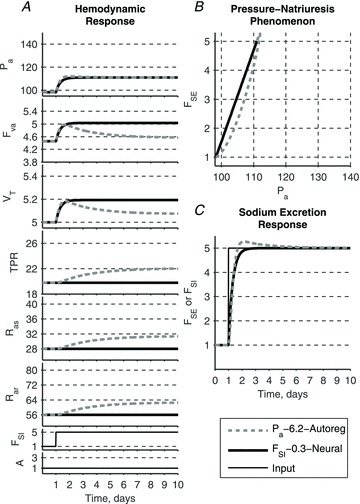
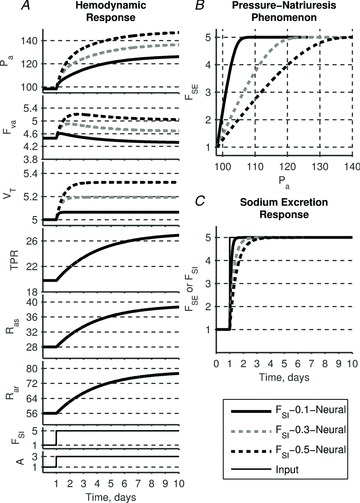
A conceptually novel mathematical model of neurogenic angiotensin II-salt hypertension is developed and analysed. The model consists of a lumped parameter circulatory model with two parallel vascular beds; two distinct control mechanisms for both natriuresis and arterial resistances can be implemented, resulting in four versions of the model. In contrast with the classical Guyton-Coleman model (GC model) of hypertension, in the standard version of our new model natriuresis is assumed to be independent of arterial pressure and instead driven solely by sodium intake; arterial resistances are driven by increased sympathetic nervous system activity in response to the elevated plasma angiotensin II and increased salt intake (AngII-salt). We compare the standard version of our new model against a simplified Guyton-Coleman model in which natriuresis is a function of arterial pressure via the pressure-natriuresis mechanism, and arterial resistances are controlled via the whole-body autoregulation mechanism. We show that the simplified GC model and the new model correctly predict haemodynamic and renal excretory responses to induced changes in angiotensin II and sodium inputs. Importantly, the new model reproduces the pressure-natriuresis relationship--the correlation between arterial pressure and sodium excretion--despite the assumption of pressure-independent natriuresis. These results show that our model provides a conceptually new alternative to Guyton's theory without contradicting observed haemodynamic changes or pressure-natriuresis relationships. Furthermore, the new model supports the view that hypertension need not necessarily have a renal aetiology and that long-term arterial pressure could be determined by sympathetic nervous system activity without involving the renal sympathetic nerves.
Figures
Comment in
-
Unsupported assumption in model of salt-sensitive hypertension.J Physiol. 2013 Jun 1;591(11):2963. doi: 10.1113/jphysiol.2013.253872. J Physiol. 2013. PMID: 23729797 Free PMC article. No abstract available.
-
Reply from V. A. Averina, H. G. Othmer, G. D. Fink and J. W. Osborn.J Physiol. 2013 Jun 1;591(11):2965. doi: 10.1113/jphysiol.2013.254607. J Physiol. 2013. PMID: 23729798 Free PMC article. No abstract available.
References
-
- Bie P. Blood volume, blood pressure and total body sodium: internal signalling and output control. Exp Physiol. 2009;195:187–196. - PubMed
-
- Caldini P, Permutt S, Waddell JA, Riley RL. Effect of epinephrine on pressure, flow, and volume relationships in the systemic circulation of dogs. Circ Res. 1974;34:606–623. - PubMed
-
- Coleman TG, Granger HJ, Guyton AC. Whole-body circulatory autoregulation and hypertension. Circ Res. 1971;28(Suppl 2):76–87. - PubMed
-
- Dorrington KL, Pandit JJ. The obligatory role of the kidney in long-term arterial blood pressure control: extending Guyton's model of the circulation. Anaesthesia. 2009;64:1218–1228. - PubMed
-
- Fine DM, Ariza-Nieto P, Osborn JW. Does whole body autoregulation mediate the hemodynamic responses to increased dietary salt in rats with clamped angiotensin II. Am J Physiol Heart Circ Physiol. 2003;285:H2670–2678. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous