

Pulmonary alveolar proteinosis
- PMID: 22891182
- PMCID: PMC3411387
- DOI: 10.1155/2012/841530
Pulmonary alveolar proteinosis
Abstract
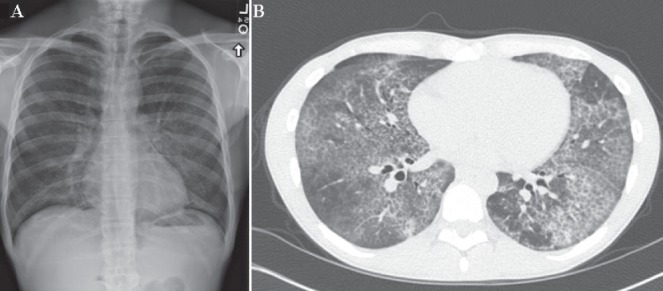
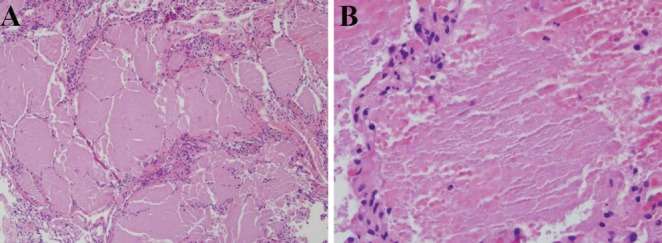
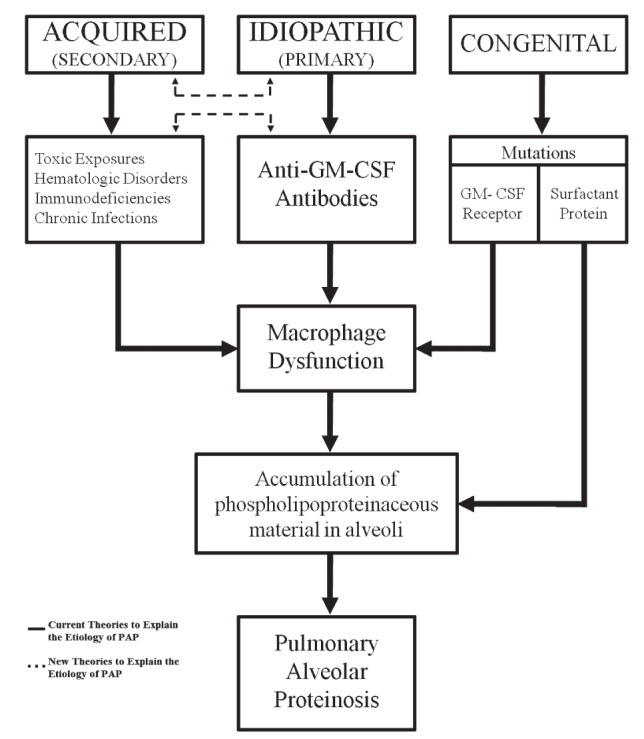
Pulmonary alveolar proteinosis (PAP) is a disease of alveolar accumulation of phospholipoproteinaceous material that results in gas exchange impairment leading to dyspnea and alveolar infiltrates. There are three forms of PAP: congenital, acquired and idiopathic; of which the latter two are predominant in the adult population. Previous case studies have found that the acquired form can be secondary to various autoimmune, infectious, malignant and environmental etiologies. Recent advances in the understanding of the pathophysiology of PAP demonstrate that the idiopathic form is due to antigranulocyte macrophage-colony stimulating factor antibodies. Therapeutic targets that replace granulocyte macrophage colony stimulating factor or remove these antibodies are being actively developed. The current standard of care is to perform whole lung lavage on these patients to clear the alveolar space to help improve respiratory physiology. A case of PAP is reported, followed by a literature review on the diagnosis and management of this rare condition with the aim of increasing awareness among physicians when treating patients who present with alveolar infiltrates.
La protéinose alvéolaire pulmonaire (PAP) se caractérise par l’accumulation alvéolaire de matière phospholipoprotéinacée qui entraîne une déficience de l’échange gazeux responsable d’une dyspnée et d’infiltrats alvéolaires. Il existe trois formes de PAP : congénitale, acquise et idiopathique. Les deux dernières prédominent dans la population adulte. Des études de cas antérieures ont déterminé que la forme acquise peut être secondaire à diverses étiologies auto-immunes, infectieuses, malignes et environnementales. Les récents progrès dans la compréhension de la physiopathologie de la PAP démontrent que la forme idiopathique découle des anticorps des facteurs de stimulation des colonies de granulocytes et de macrophages. On est à mettre au point des cibles thérapeutiques qui remplacent le facteur de stimulation des colonies de granulocytes et de macrophages ou qui suppriment ces anticorps. La norme actuelle des soins consiste à exécuter un lavage pulmonaire complet afin de dégager l’espace alvéolaire pour améliorer la physiologie respiratoire. Un cas de PAP est déclaré, suivi d’une analyse bibliographique du diagnostic et de la prise en charge de cette pathologie rare, afin de mieux y sensibiliser les médecins lorsqu’ils traitent des patients qui présentent des infiltrats alvéolaires.
Figures
References
-
- Rosen SH, Castleman B, Liebow AA. Pulmonary alveolar proteinosis. N Engl J Med. 1958;258:1123–42. - PubMed
-
- Hunt S, Miller AL, Schissel S, Ross JJ. A crazy cause of dyspnea. N Engl J Med. 2010;363:e38. - PubMed
-
- Costabel U, Nakata K. Pulmonary alveolar proteinosis associated with dust inhalation: Not secondary but autoimmune? Am J Respir Crit Care Med. 2010;181:427–8. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
