

A series of N-terminal epitope tagged Hdh knock-in alleles expressing normal and mutant huntingtin: their application to understanding the effect of increasing the length of normal Huntingtin's polyglutamine stretch on CAG140 mouse model pathogenesis
- PMID: 22892315
- PMCID: PMC3499431
- DOI: 10.1186/1756-6606-5-28
A series of N-terminal epitope tagged Hdh knock-in alleles expressing normal and mutant huntingtin: their application to understanding the effect of increasing the length of normal Huntingtin's polyglutamine stretch on CAG140 mouse model pathogenesis
Abstract
Background: Huntington's disease (HD) is an autosomal dominant neurodegenerative disease that is caused by the expansion of a polyglutamine (polyQ) stretch within Huntingtin (htt), the protein product of the HD gene. Although studies in vitro have suggested that the mutant htt can act in a potentially dominant negative fashion by sequestering wild-type htt into insoluble protein aggregates, the role of the length of the normal htt polyQ stretch, and the adjacent proline-rich region (PRR) in modulating HD mouse model pathogenesis is currently unknown.
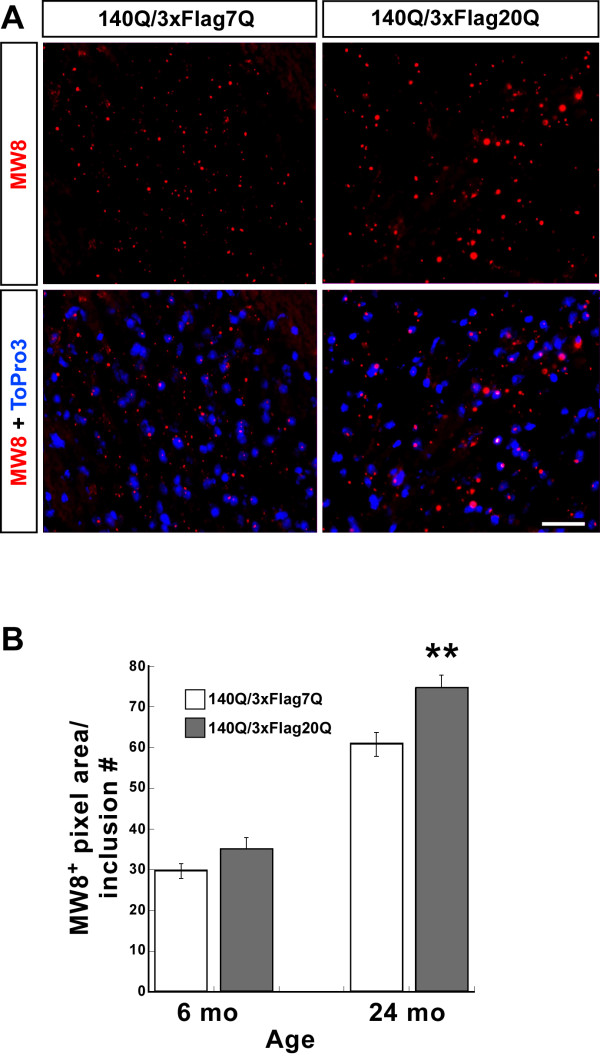
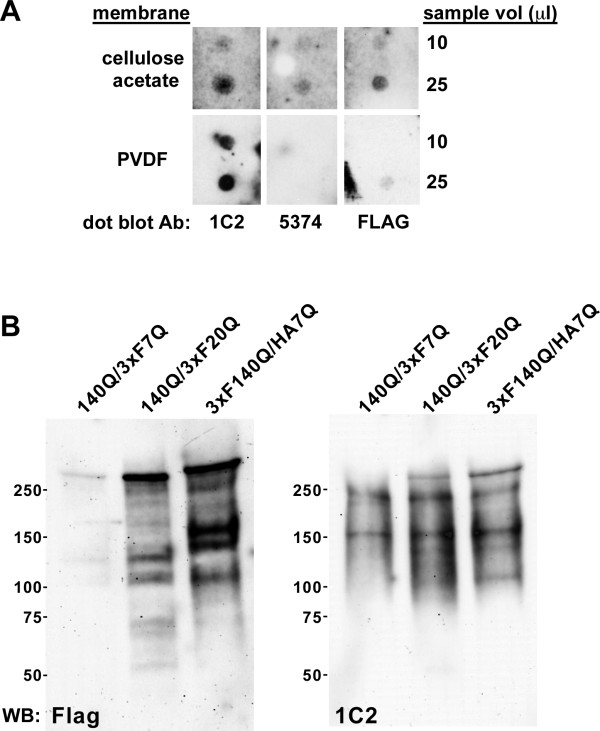
Results: We describe the generation and characterization of a series of knock-in HD mouse models that express versions of the mouse HD gene (Hdh) encoding N-terminal hemaglutinin (HA) or 3xFlag epitope tagged full-length htt with different polyQ lengths (HA7Q-, 3xFlag7Q-, 3xFlag20Q-, and 3xFlag140Q-htt) and substitution of the adjacent mouse PRR with the human PRR (3xFlag20Q- and 3xFlag140Q-htt). Using co-immunoprecipitation and immunohistochemistry analyses, we detect no significant interaction between soluble full-length normal 7Q- htt and mutant (140Q) htt, but we do observe N-terminal fragments of epitope-tagged normal htt in mutant htt aggregates. When the sequences encoding normal mouse htt's polyQ stretch and PRR are replaced with non-pathogenic human sequence in mice also expressing 140Q-htt, aggregation foci within the striatum, and the mean size of htt inclusions are increased, along with an increase in striatal lipofuscin and gliosis.
Conclusion: In mice, soluble full-length normal and mutant htt are predominantly monomeric. In heterozygous knock-in HD mouse models, substituting the normal mouse polyQ and PRR with normal human sequence can exacerbate some neuropathological phenotypes.
Figures









Similar articles
-
Deletion of the huntingtin polyglutamine stretch enhances neuronal autophagy and longevity in mice.PLoS Genet. 2010 Feb 5;6(2):e1000838. doi: 10.1371/journal.pgen.1000838. PLoS Genet. 2010. PMID: 20140187 Free PMC article.
-
Generation and Characterization of Knock-in Mouse Models Expressing Versions of Huntingtin with Either an N17 or a Combined PolyQ and Proline-Rich Region Deletion.J Huntingtons Dis. 2017;6(1):47-62. doi: 10.3233/JHD-160231. J Huntingtons Dis. 2017. PMID: 28211815 Free PMC article.
-
Multiple phenotypes in Huntington disease mouse neural stem cells.Mol Cell Neurosci. 2012 May;50(1):70-81. doi: 10.1016/j.mcn.2012.03.011. Epub 2012 Apr 6. Mol Cell Neurosci. 2012. PMID: 22508027 Free PMC article.
-
Selective degeneration in YAC mouse models of Huntington disease.Brain Res Bull. 2007 Apr 30;72(2-3):124-31. doi: 10.1016/j.brainresbull.2006.10.018. Epub 2006 Nov 16. Brain Res Bull. 2007. PMID: 17352936 Review.
-
Polyglutamine pathogenesis.Philos Trans R Soc Lond B Biol Sci. 1999 Jun 29;354(1386):1005-11. doi: 10.1098/rstb.1999.0452. Philos Trans R Soc Lond B Biol Sci. 1999. PMID: 10434299 Free PMC article. Review.
Cited by
-
Mutant Huntingtin Protein Interaction Map Implicates Dysregulation of Multiple Cellular Pathways in Neurodegeneration of Huntington's Disease.J Huntingtons Dis. 2022;11(3):243-267. doi: 10.3233/JHD-220538. J Huntingtons Dis. 2022. PMID: 35871359 Free PMC article.
-
Recent Microscopy Advances and the Applications to Huntington's Disease Research.J Huntingtons Dis. 2022;11(3):269-280. doi: 10.3233/JHD-220536. J Huntingtons Dis. 2022. PMID: 35848031 Free PMC article. Review.
-
A large scale Huntingtin protein interaction network implicates Rho GTPase signaling pathways in Huntington disease.J Biol Chem. 2014 Mar 7;289(10):6709-6726. doi: 10.1074/jbc.M113.523696. Epub 2014 Jan 9. J Biol Chem. 2014. PMID: 24407293 Free PMC article.
-
Multi-epitope immunocapture of huntingtin reveals striatum-selective molecular signatures.Mol Syst Biol. 2025 May;21(5):492-522. doi: 10.1038/s44320-025-00096-3. Epub 2025 Apr 1. Mol Syst Biol. 2025. PMID: 40169779 Free PMC article.
-
Dynamics of huntingtin protein interactions in the striatum identifies candidate modifiers of Huntington disease.Cell Syst. 2022 Apr 20;13(4):304-320.e5. doi: 10.1016/j.cels.2022.01.005. Epub 2022 Feb 10. Cell Syst. 2022. PMID: 35148841 Free PMC article.
References
-
- Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/S0092-8674(00)80513-9. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
