

Upregulated function of mitochondria-associated ER membranes in Alzheimer disease
- PMID: 22892566
- PMCID: PMC3492725
- DOI: 10.1038/emboj.2012.202
Upregulated function of mitochondria-associated ER membranes in Alzheimer disease
Abstract
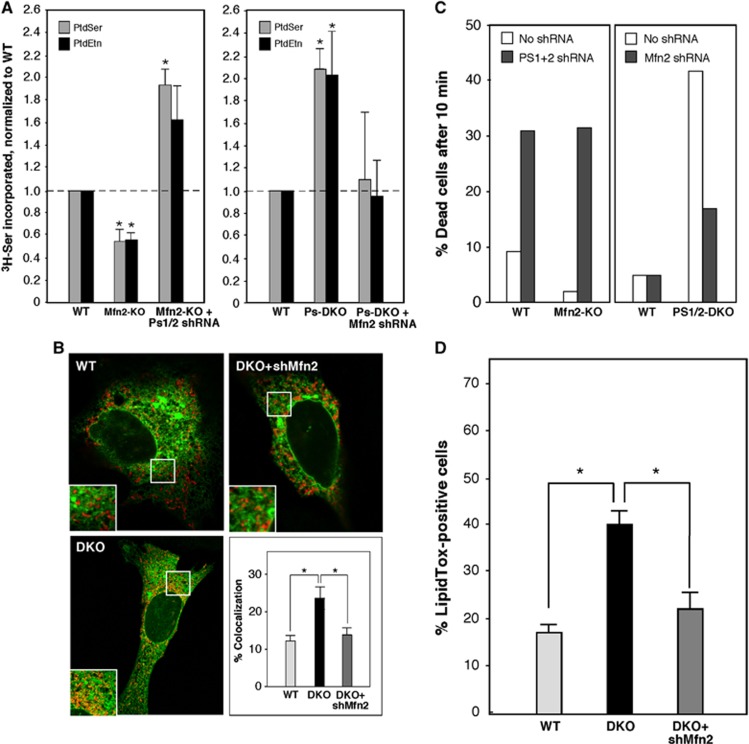
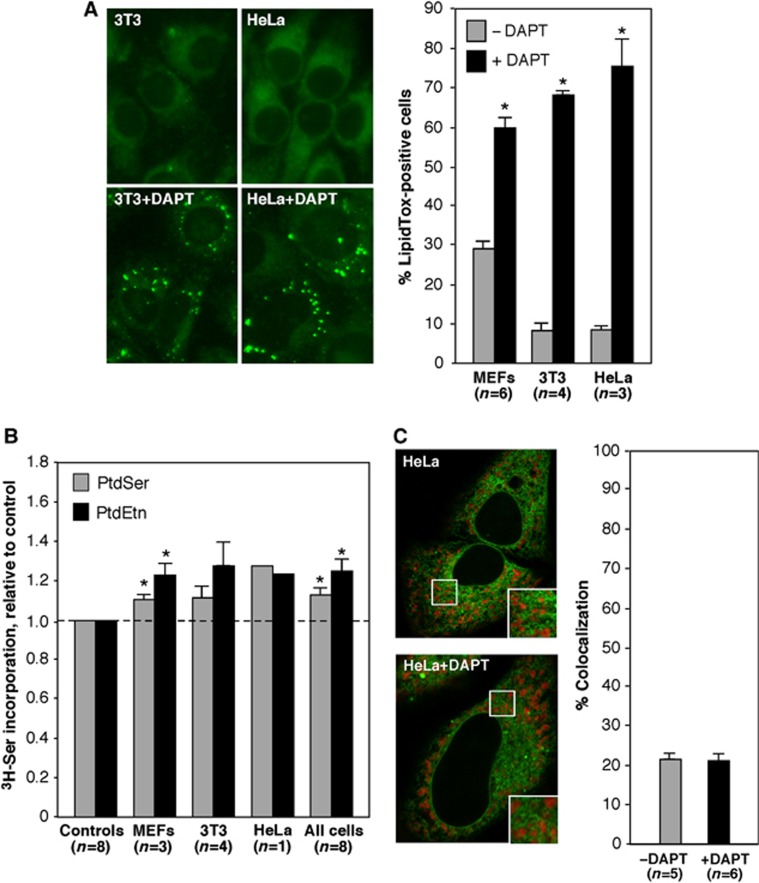
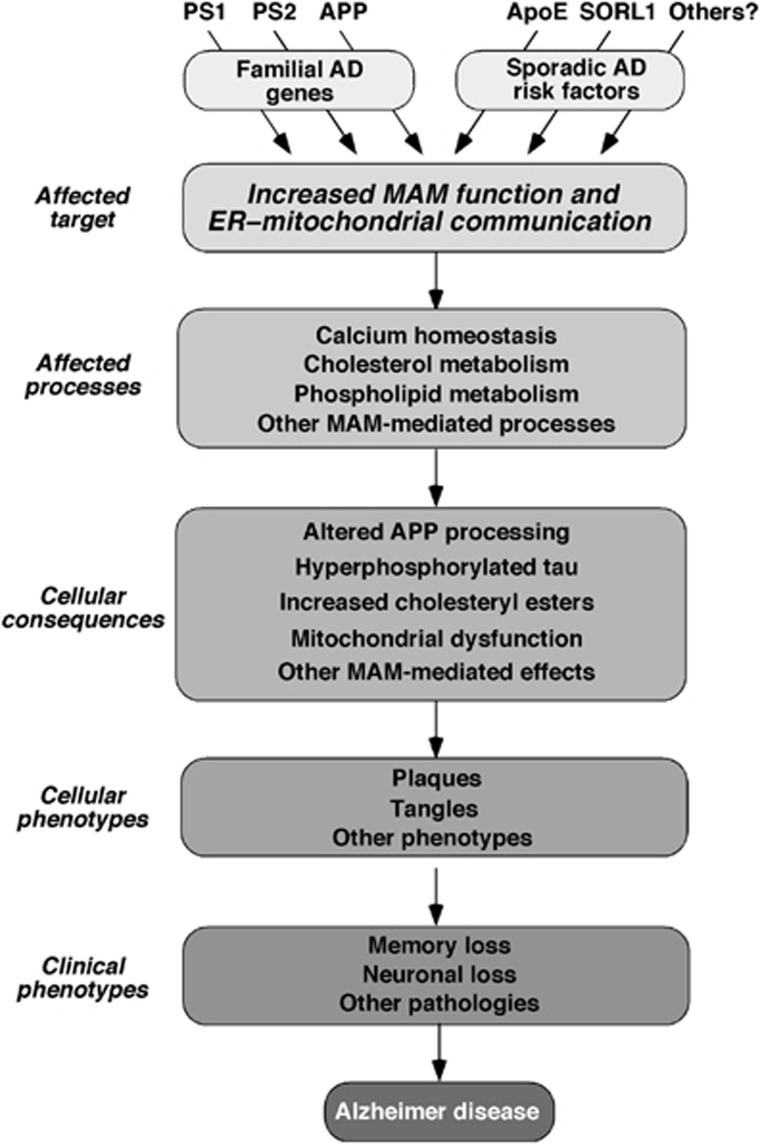
Alzheimer disease (AD) is associated with aberrant processing of the amyloid precursor protein (APP) by γ-secretase, via an unknown mechanism. We recently showed that presenilin-1 and -2, the catalytic components of γ-secretase, and γ-secretase activity itself, are highly enriched in a subcompartment of the endoplasmic reticulum (ER) that is physically and biochemically connected to mitochondria, called mitochondria-associated ER membranes (MAMs). We now show that MAM function and ER-mitochondrial communication-as measured by cholesteryl ester and phospholipid synthesis, respectively-are increased significantly in presenilin-mutant cells and in fibroblasts from patients with both the familial and sporadic forms of AD. We also show that MAM is an intracellular detergent-resistant lipid raft (LR)-like domain, consistent with the known presence of presenilins and γ-secretase activity in rafts. These findings may help explain not only the aberrant APP processing but also a number of other biochemical features of AD, including altered lipid metabolism and calcium homeostasis. We propose that upregulated MAM function at the ER-mitochondrial interface, and increased cross-talk between these two organelles, may play a hitherto unrecognized role in the pathogenesis of AD.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
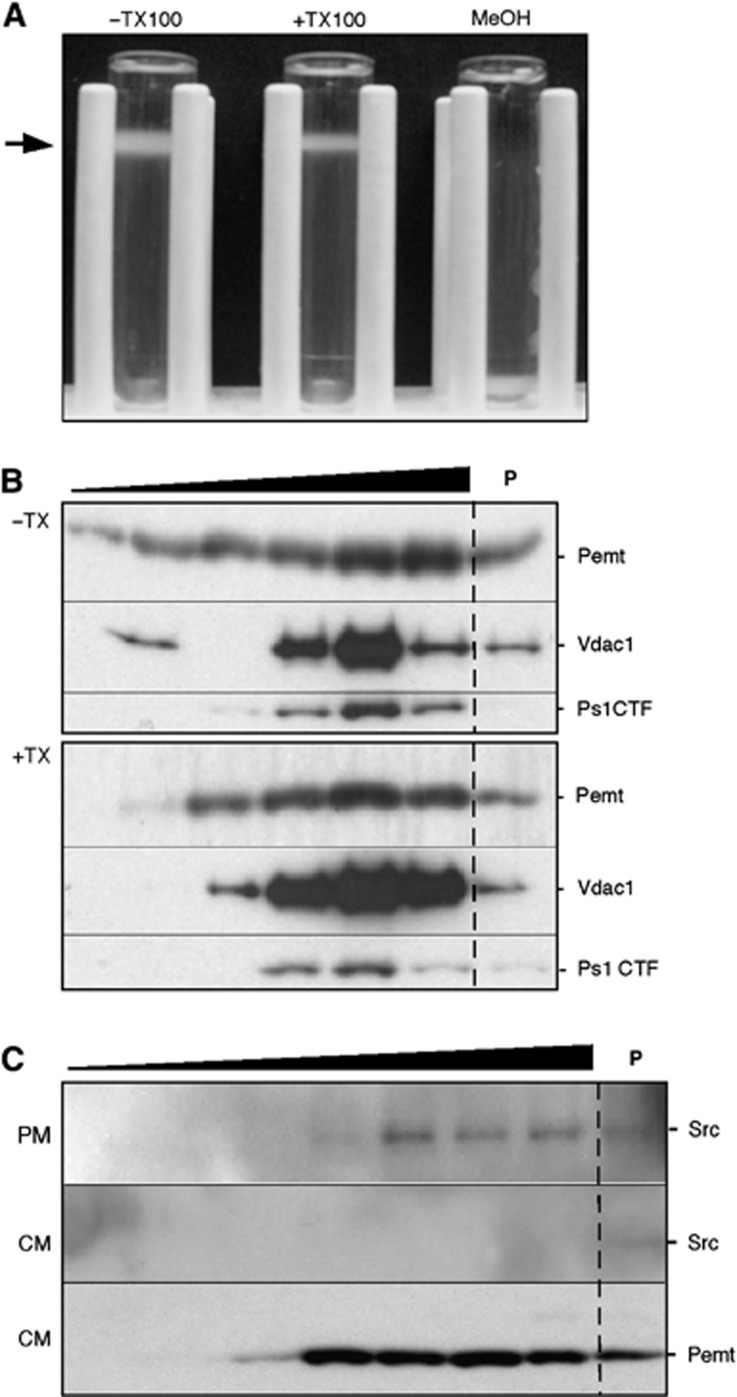
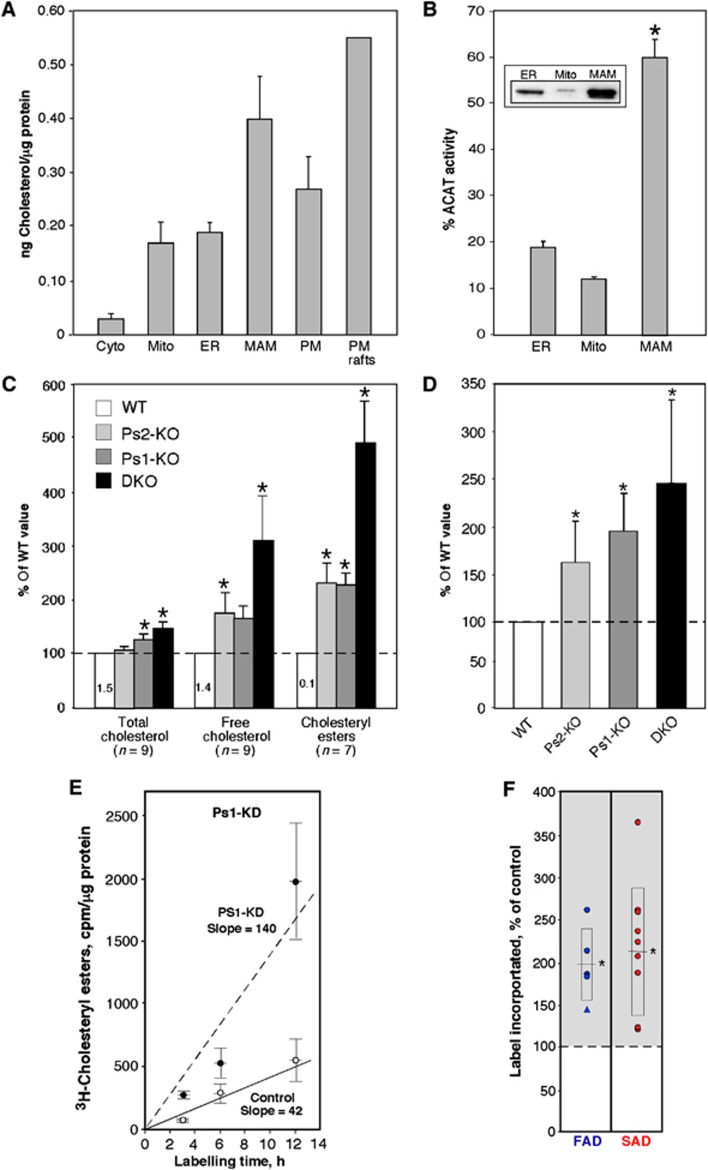
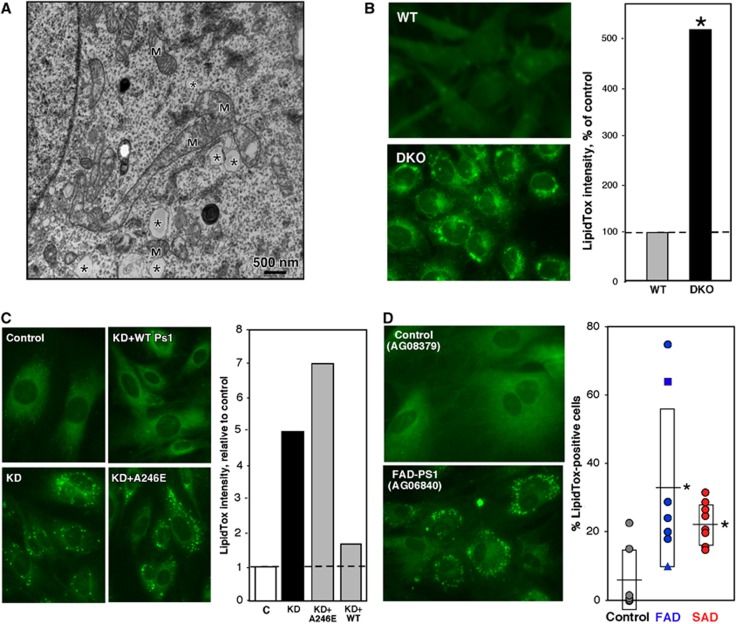
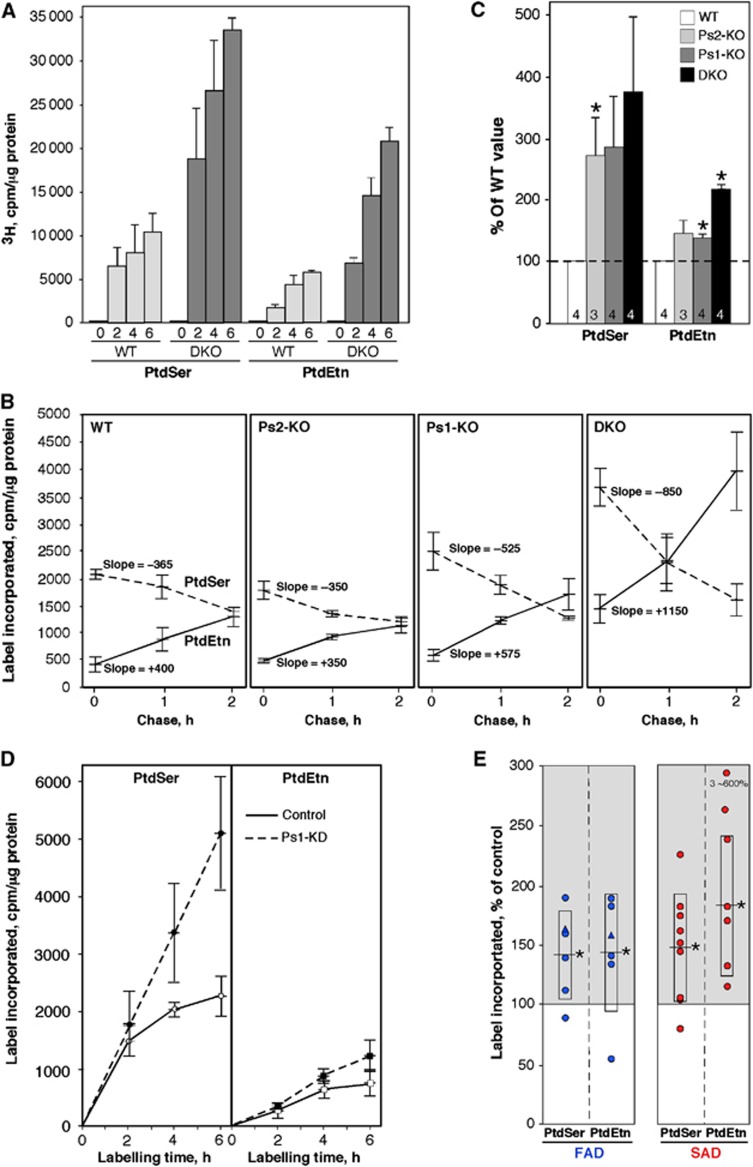
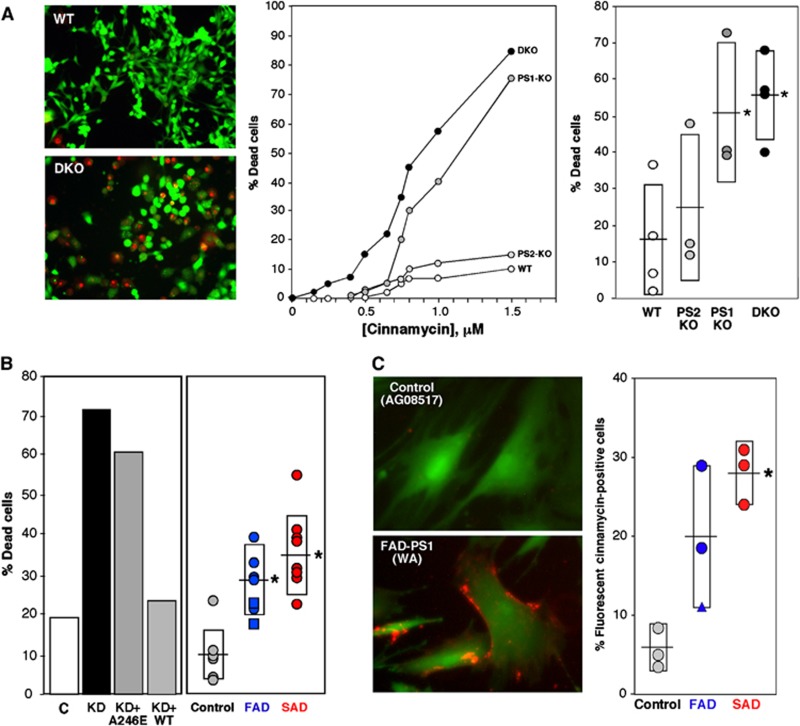
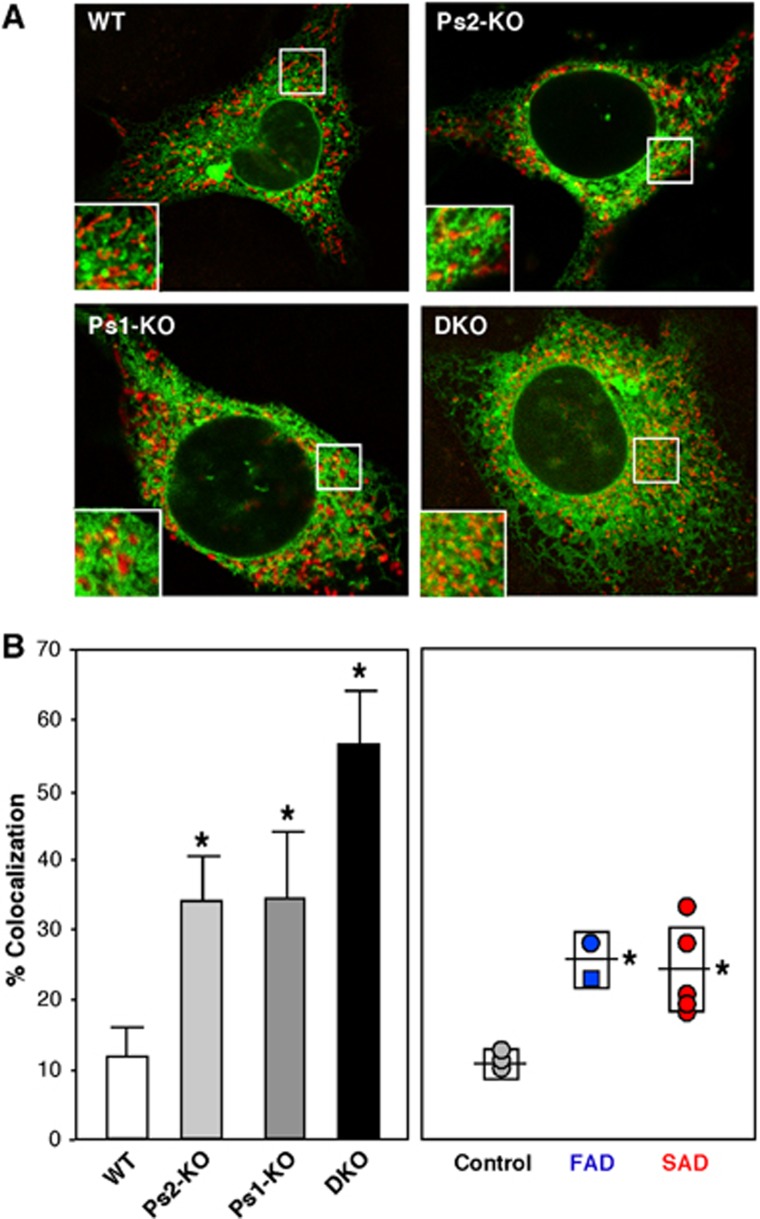
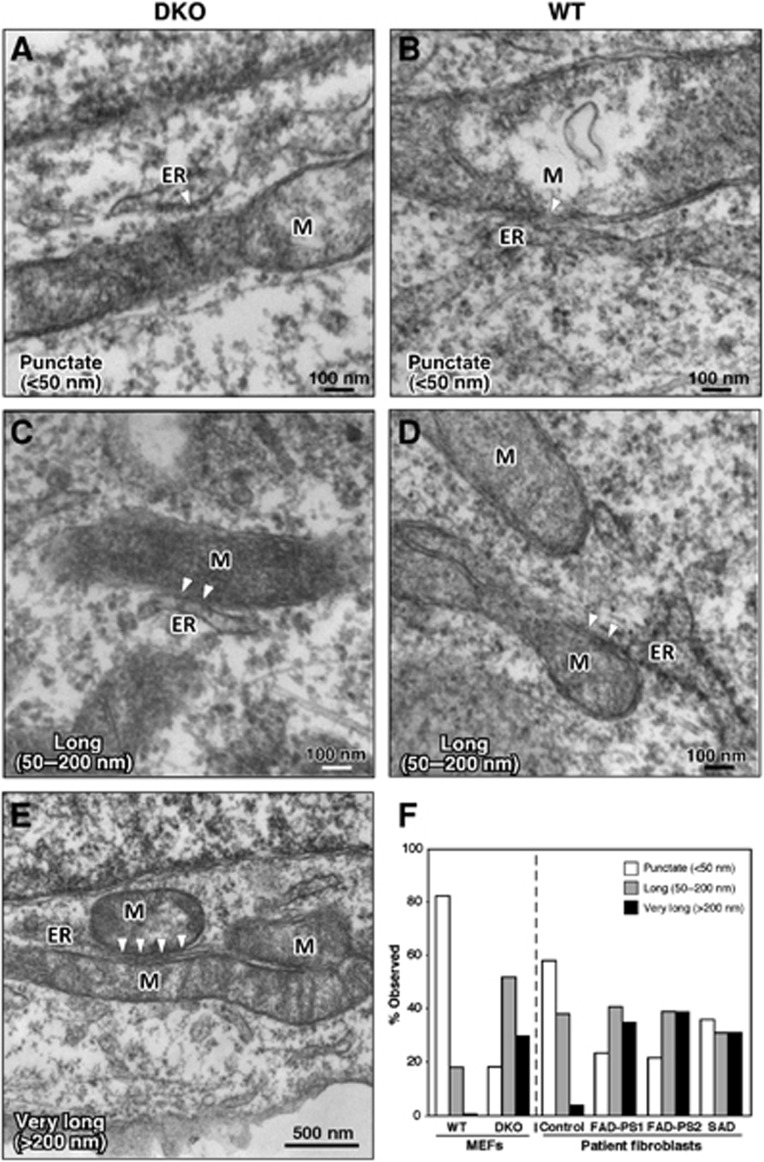
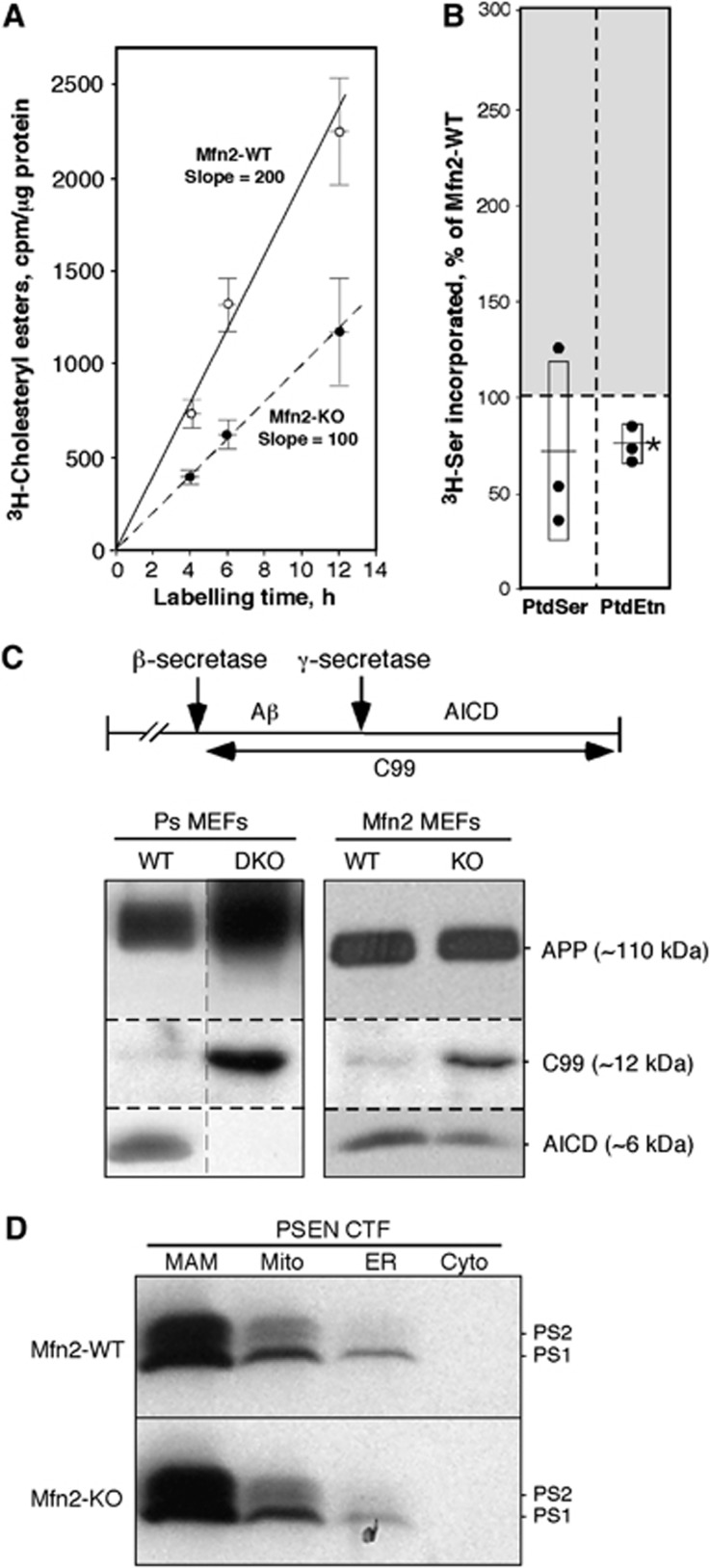
Comment in
-
Close encounter: mitochondria, endoplasmic reticulum and Alzheimer's disease.EMBO J. 2012 Nov 5;31(21):4095-7. doi: 10.1038/emboj.2012.279. Epub 2012 Oct 9. EMBO J. 2012. PMID: 23047154 Free PMC article.
References
-
- Ankarcrona M, Hultenby K (2002) Presenilin-1 is located in rat mitochondria. Biochem Biophys Res Commun 295: 766–770 - PubMed
-
- Annaert WG, Levesque L, Craessaerts K, Dierinck I, Snellings G, Westaway D, St George-Hyslop P, Cordell B, Fraser P, De Strooper B (1999) Presenilin 1 controls γ-secretase processing of amyloid precursor protein in pre-golgi compartments of hippocampal neurons. J Cell Biol 147: 277–294 - PMC - PubMed
-
- Bentahir M, Nyabi O, Verhamme J, Tolia A, Horre K, Wiltfang J, Esselmann H, De Strooper B (2006) Presenilin clinical mutations can affect γ-secretase activity by different mechanisms. J Neurochem 96: 732–742 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
