

Dissecting sources of quantitative gene expression pattern divergence between Drosophila species
- PMID: 22893002
- PMCID: PMC3435502
- DOI: 10.1038/msb.2012.35
Dissecting sources of quantitative gene expression pattern divergence between Drosophila species
Abstract
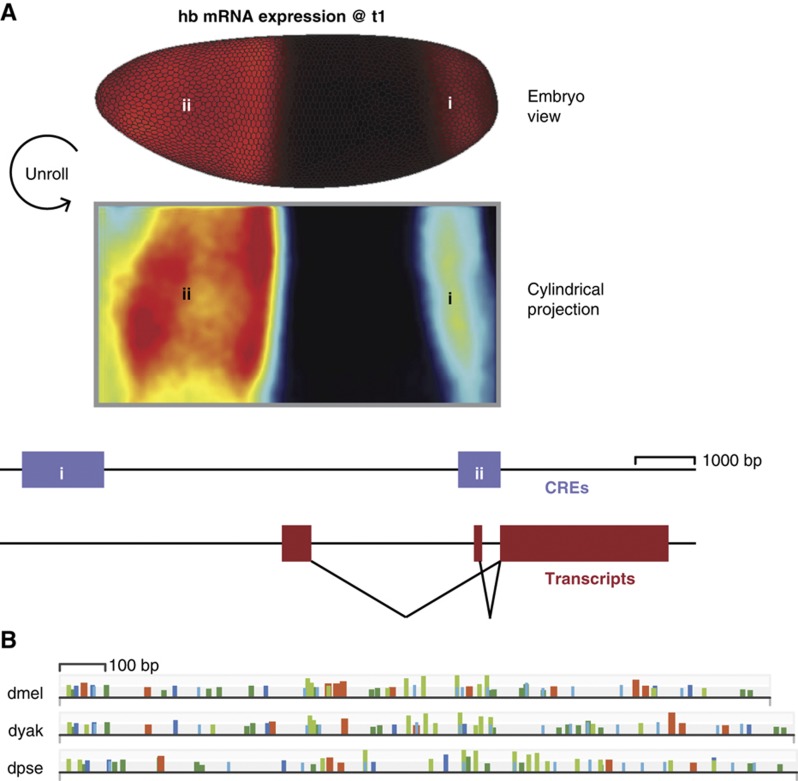
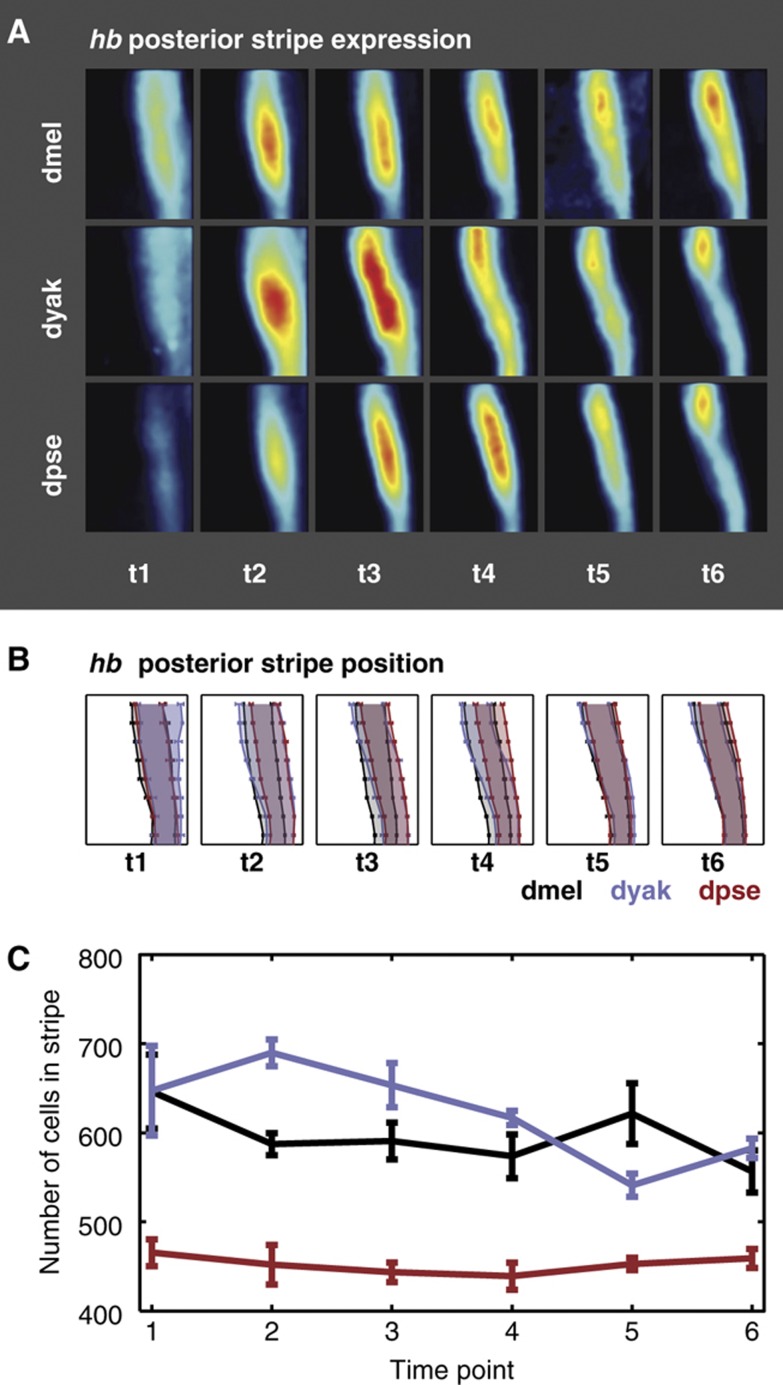
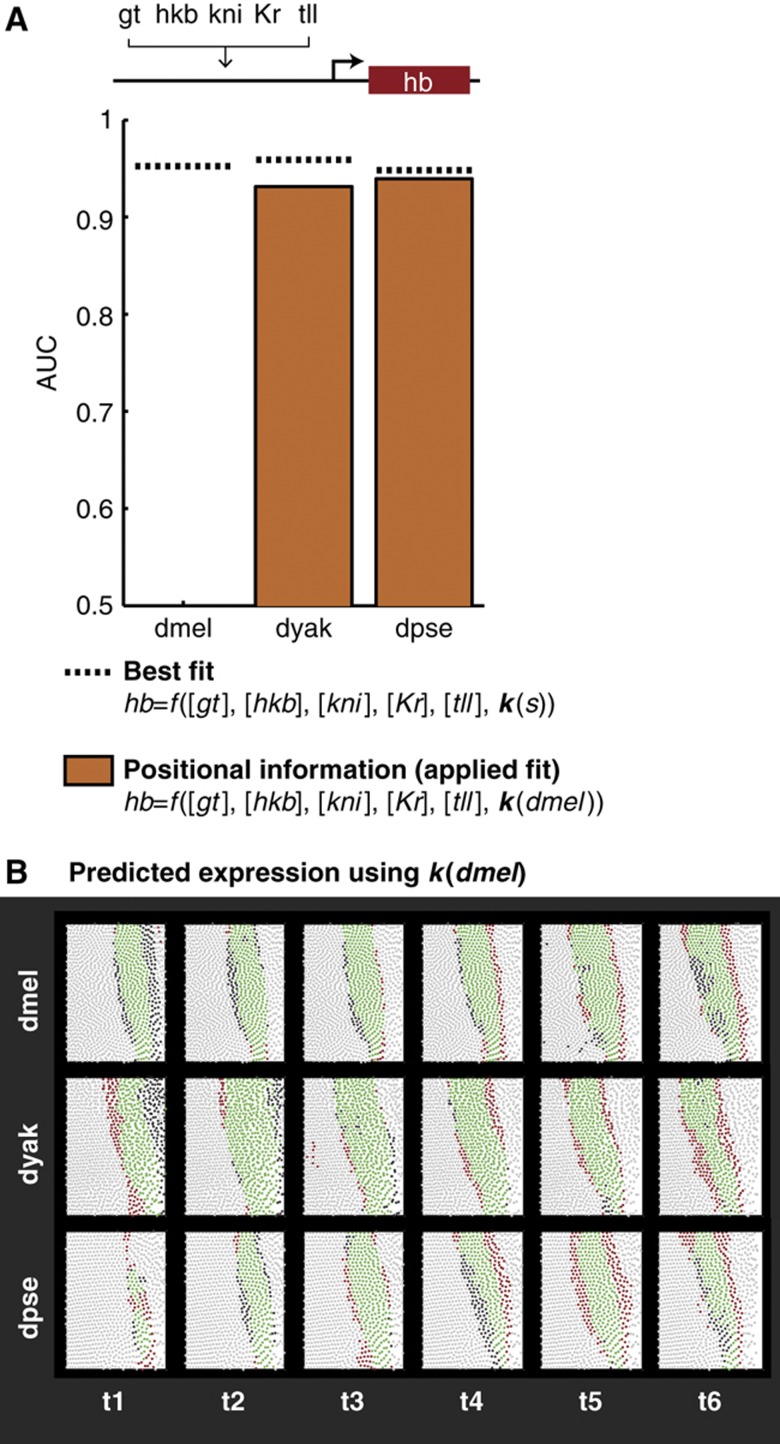
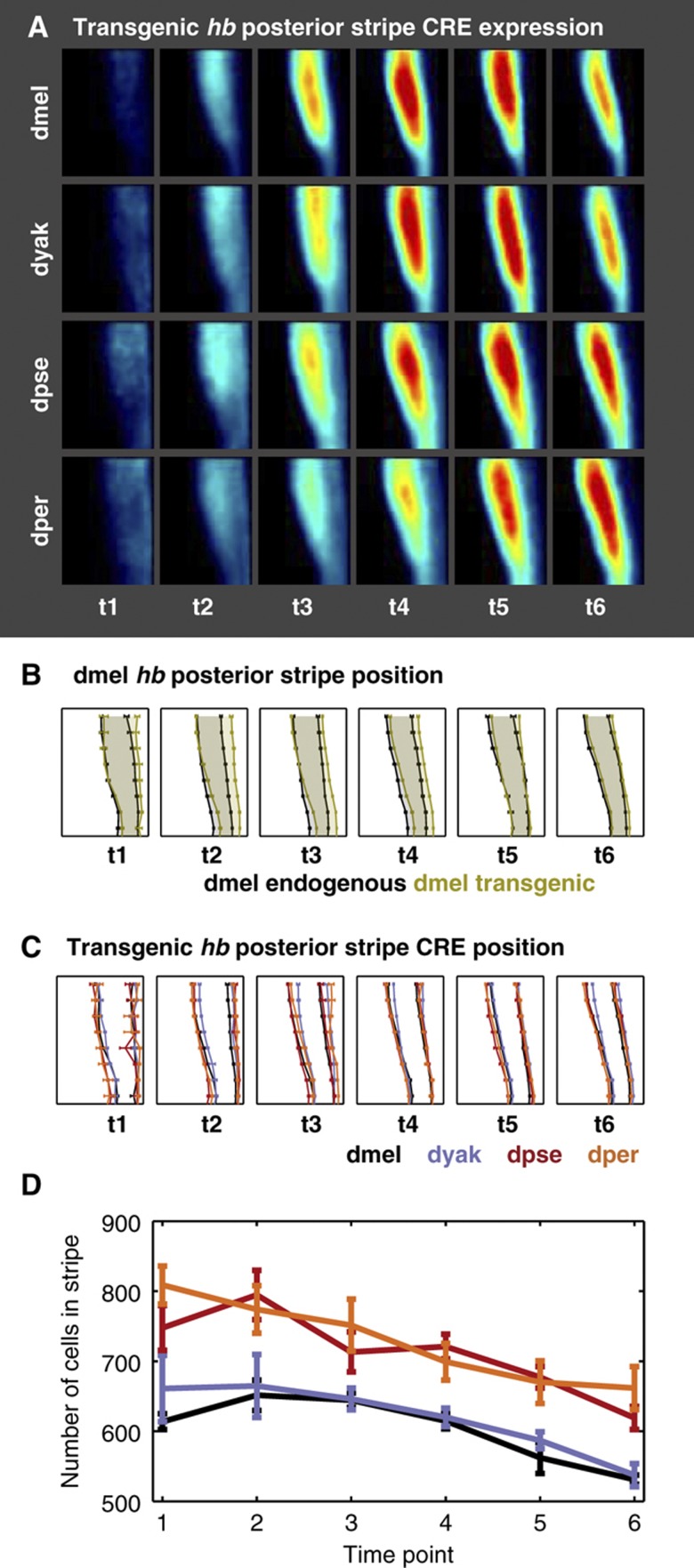
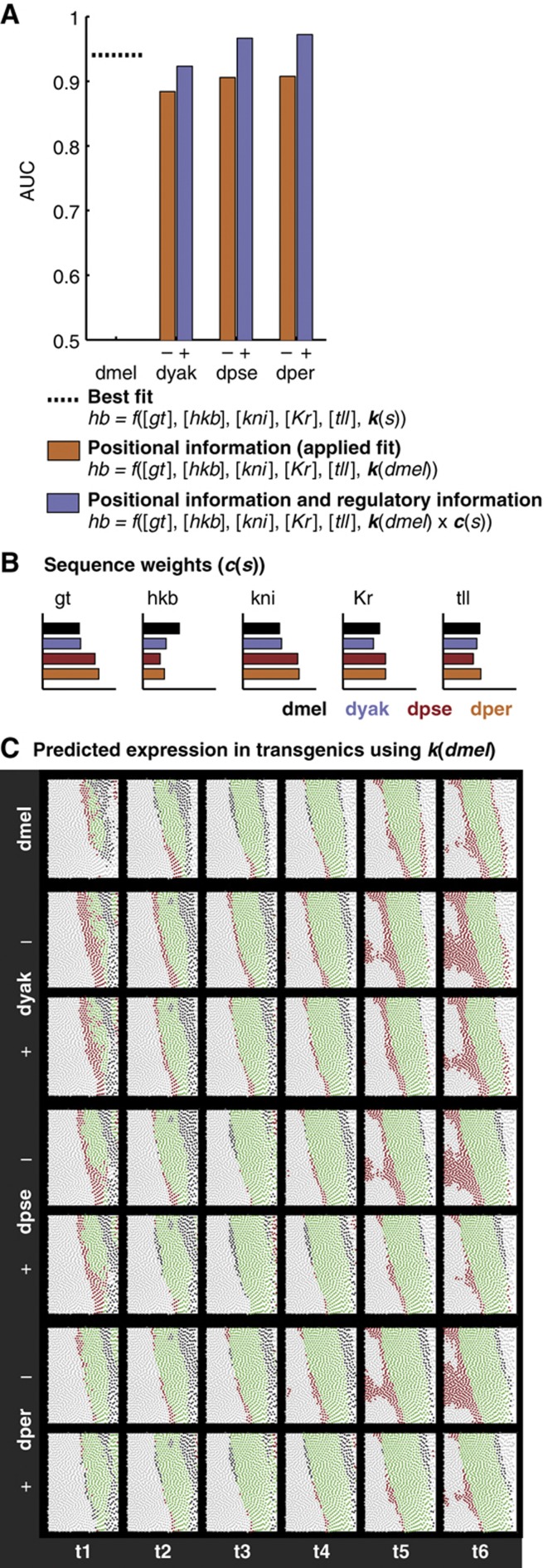
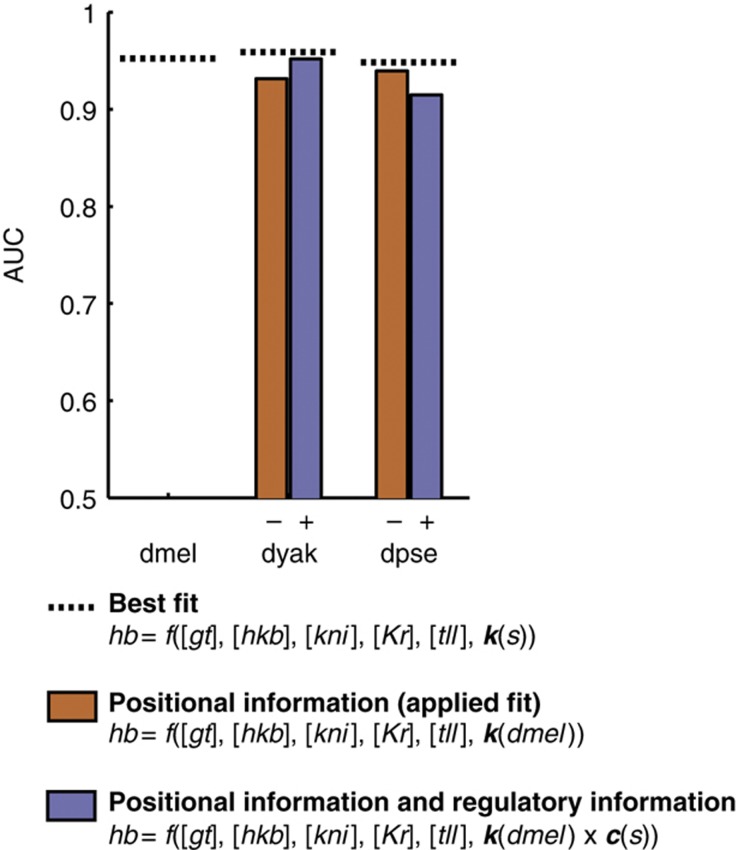
Gene expression patterns can diverge between species due to changes in a gene's regulatory DNA or changes in the proteins, e.g., transcription factors (TFs), that regulate the gene. We developed a modeling framework to uncover the sources of expression differences in blastoderm embryos of three Drosophila species, focusing on the regulatory circuit controlling expression of the hunchback (hb) posterior stripe. Using this framework and cellular-resolution expression measurements of hb and its regulating TFs, we found that changes in the expression patterns of hb's TFs account for much of the expression divergence. We confirmed our predictions using transgenic D. melanogaster lines, which demonstrate that this set of orthologous cis-regulatory elements (CREs) direct similar, but not identical, expression patterns. We related expression pattern differences to sequence changes in the CRE using a calculation of the CRE's TF binding site content. By applying this calculation in both the transgenic and endogenous contexts, we found that changes in binding site content affect sensitivity to regulating TFs and that compensatory evolution may occur in circuit components other than the CRE.
Figures
References
-
- Ardehali MB, Lis JT (2009) Tracking rates of transcription and splicing in vivo. Nat Struct Mol Biol 16: 1123–1124 - PubMed
-
- Arnosti DN, Barolo S, Levine M, Small S (1996) The eve stripe 2 enhancer employs multiple modes of transcriptional synergy. Development 122: 205–214 - PubMed
-
- Ben-Tabou de-Leon S, Davidson EH (2007) Gene regulation: gene control network in development. Annu Rev Biophys Biomol Struct 36: 191. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
