

Genetic characterization in symptomatic female DMD carriers: lack of relationship between X-inactivation, transcriptional DMD allele balancing and phenotype
- PMID: 22894145
- PMCID: PMC3459813
- DOI: 10.1186/1471-2350-13-73
Genetic characterization in symptomatic female DMD carriers: lack of relationship between X-inactivation, transcriptional DMD allele balancing and phenotype
Abstract
Background: Although Duchenne and Becker muscular dystrophies, X-linked recessive myopathies, predominantly affect males, a clinically significant proportion of females manifesting symptoms have also been reported. They represent an heterogeneous group characterized by variable degrees of muscle weakness and/or cardiac involvement. Though preferential inactivation of the normal X chromosome has long been considered the principal mechanism behind disease manifestation in these females, supporting evidence is controversial.
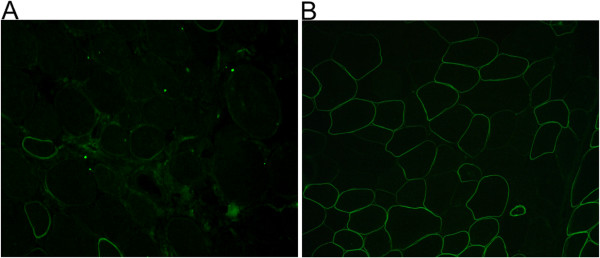
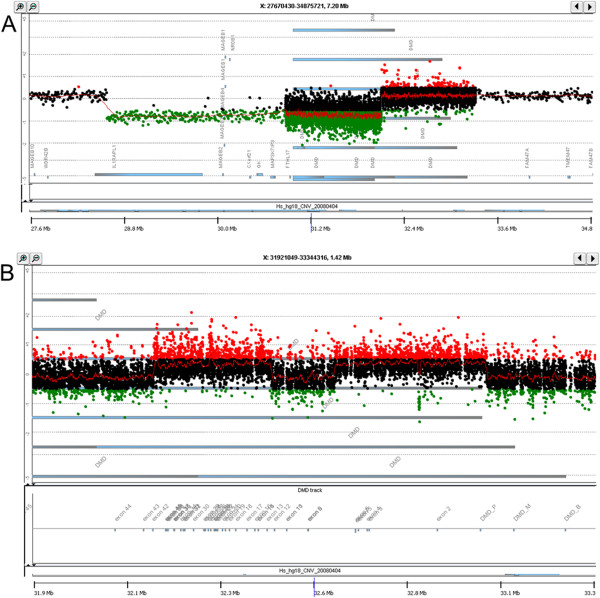
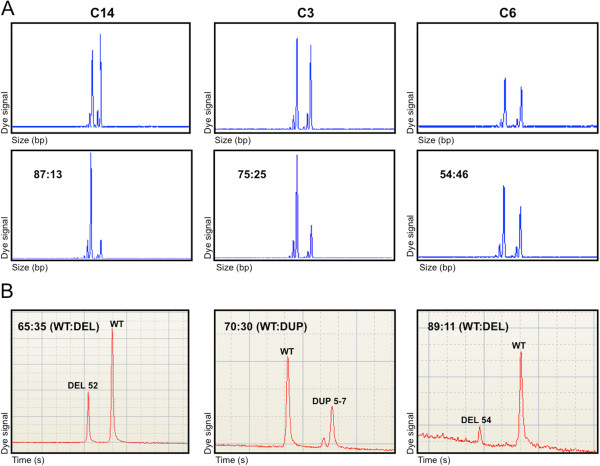
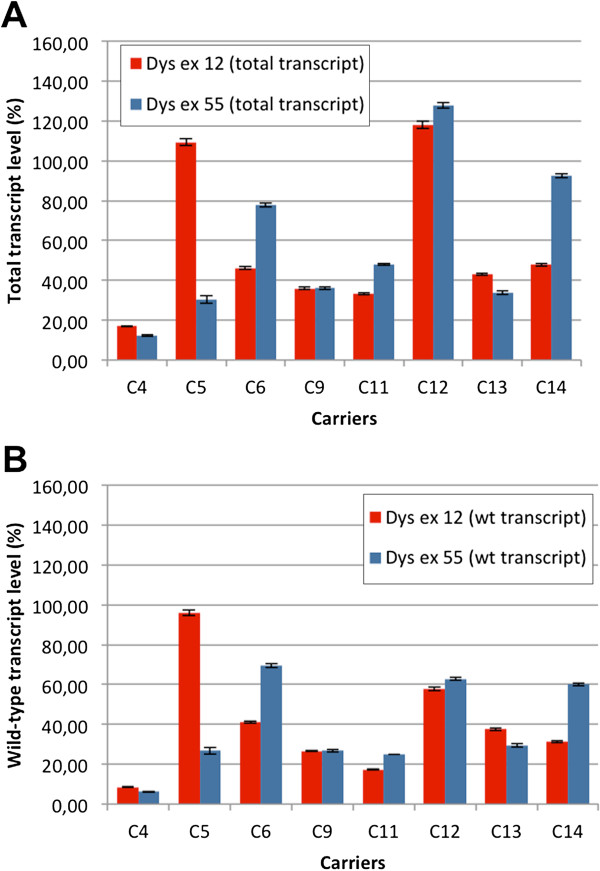
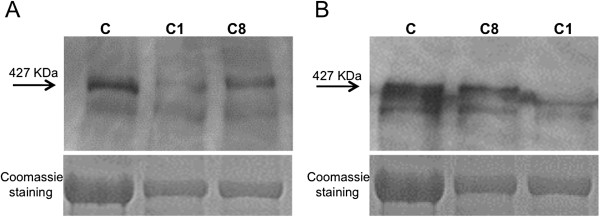
Methods: Eighteen females showing a mosaic pattern of dystrophin expression on muscle biopsy were recruited and classified as symptomatic (7) or asymptomatic (11), based on the presence or absence of muscle weakness. The causative DMD gene mutations were identified in all cases, and the X-inactivation pattern was assessed in muscle DNA. Transcriptional analysis in muscles was performed in all females, and relative quantification of wild-type and mutated transcripts was also performed in 9 carriers. Dystrophin protein was quantified by immunoblotting in 2 females.
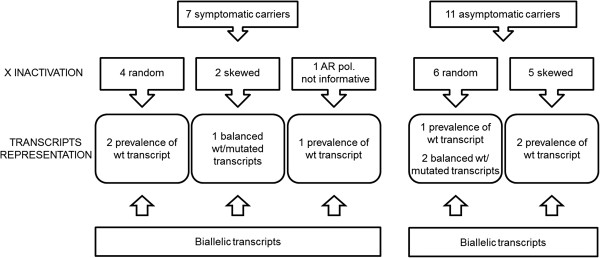
Results: The study highlighted a lack of relationship between dystrophic phenotype and X-inactivation pattern in females; skewed X-inactivation was found in 2 out of 6 symptomatic carriers and in 5 out of 11 asymptomatic carriers. All females were characterized by biallelic transcription, but no association was found between X-inactivation pattern and allele transcriptional balancing. Either a prevalence of wild-type transcript or equal proportions of wild-type and mutated RNAs was observed in both symptomatic and asymptomatic females. Moreover, very similar levels of total and wild-type transcripts were identified in the two groups of carriers.
Conclusions: This is the first study deeply exploring the DMD transcriptional behaviour in a cohort of female carriers. Notably, no relationship between X-inactivation pattern and transcriptional behaviour of DMD gene was observed, suggesting that the two mechanisms are regulated independently. Moreover, neither the total DMD transcript level, nor the relative proportion of the wild-type transcript do correlate with the symptomatic phenotype.
Figures
References
-
- Moser H, Emery AE. The manifesting carrier in Duchenne muscular dystrophy. Clin Genet. 1974;5(4):271–284. - PubMed
-
- Norman A, Harper P. A survey of manifesting carriers of Duchenne and Becker muscular dystrophy in Wales. Clin Genet. 1989;36(1):31–37. - PubMed
-
- Hoogerwaard EM, Bakker E, Ippel PF, Oosterwijk JC, Majoor-Krakauer DF, Leschot NJ, Van Essen AJ, Brunner HG, van der Wouw PA, Wilde AA, de Visser M. Signs and symptoms of Duchenne muscular dystrophy and Becker muscular dystrophy among carriers in the Netherlands: a cohort study. Lancet. 1999;353:2116–2119. doi: 10.1016/S0140-6736(98)10028-4. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
