

Opioid activation of toll-like receptor 4 contributes to drug reinforcement
- PMID: 22895704
- PMCID: PMC3454463
- DOI: 10.1523/JNEUROSCI.0684-12.2012
Opioid activation of toll-like receptor 4 contributes to drug reinforcement
Abstract
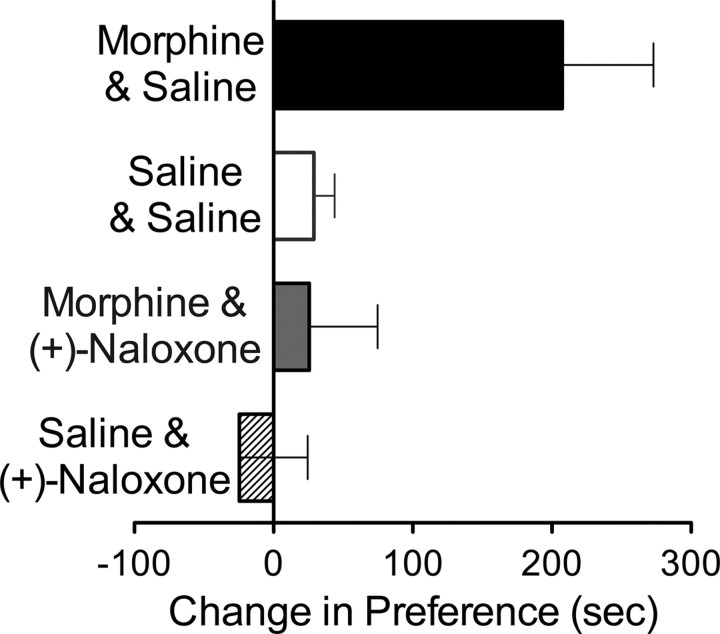
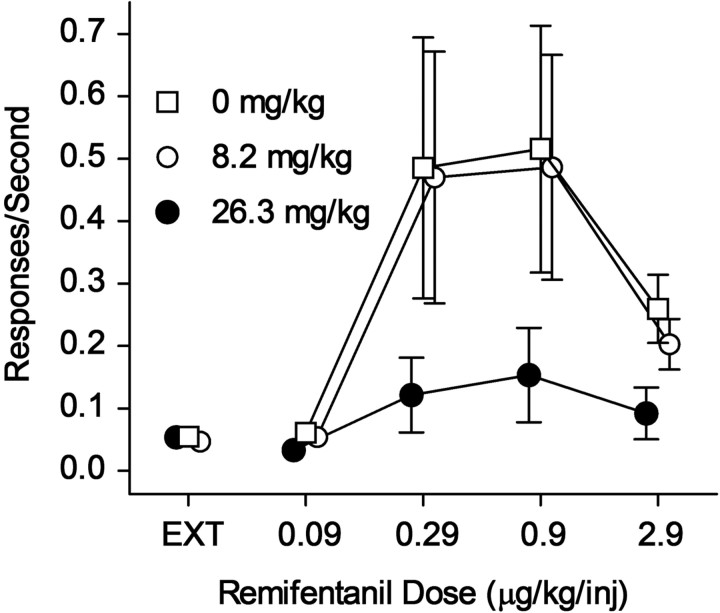
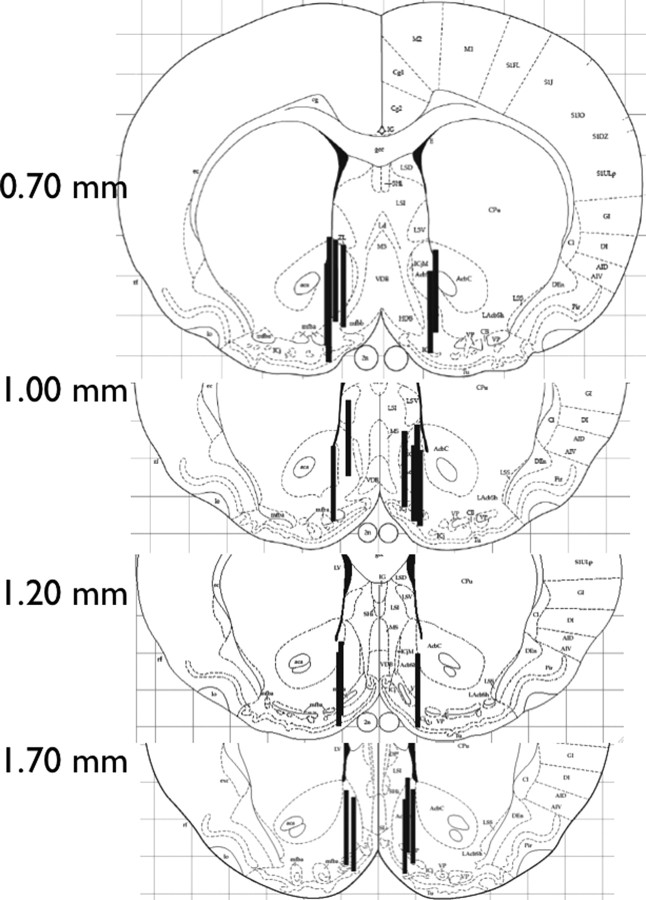
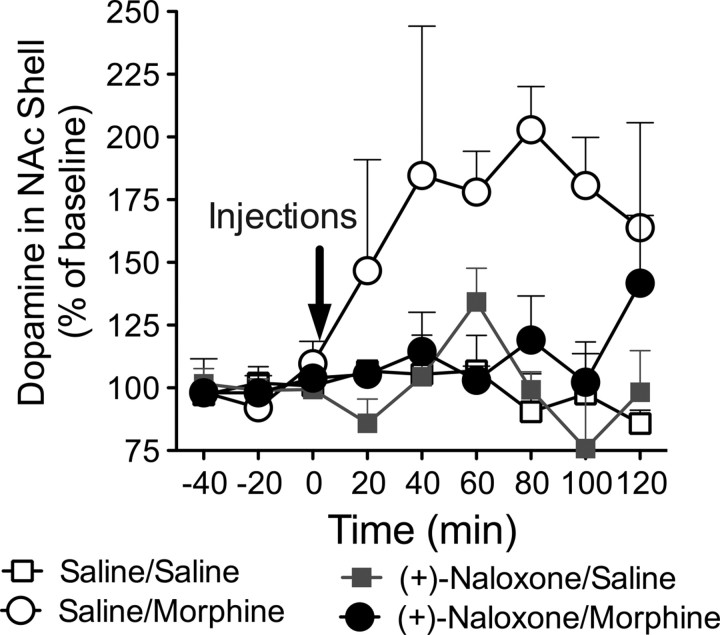
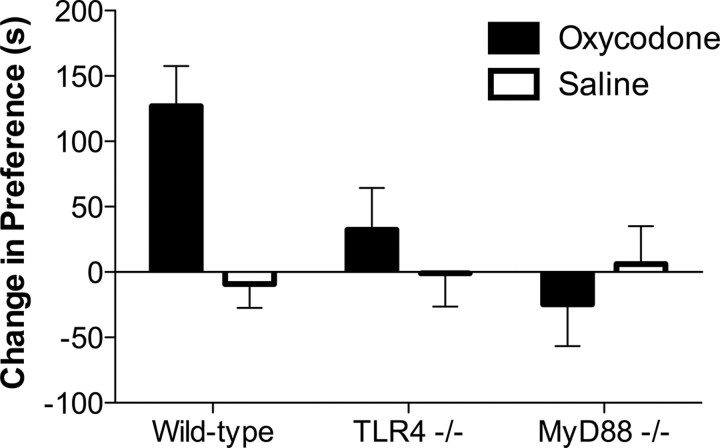
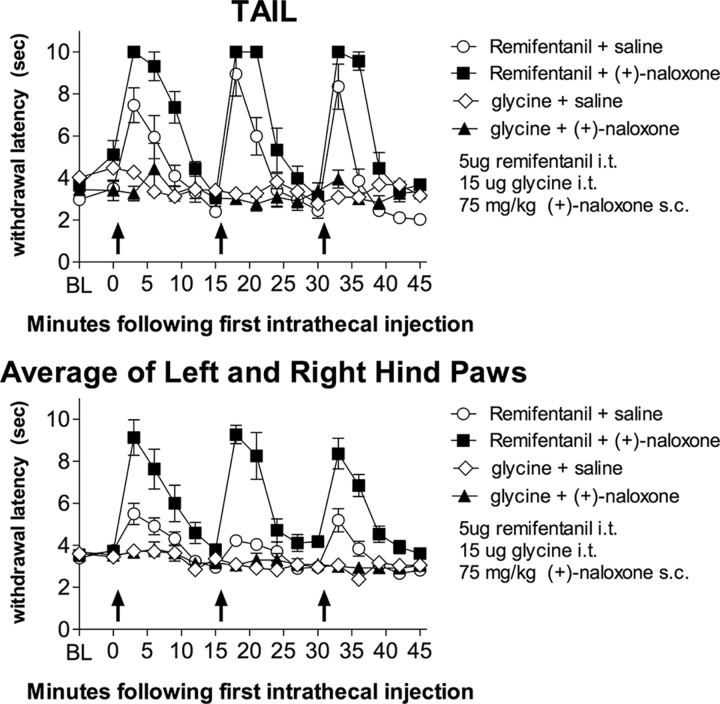
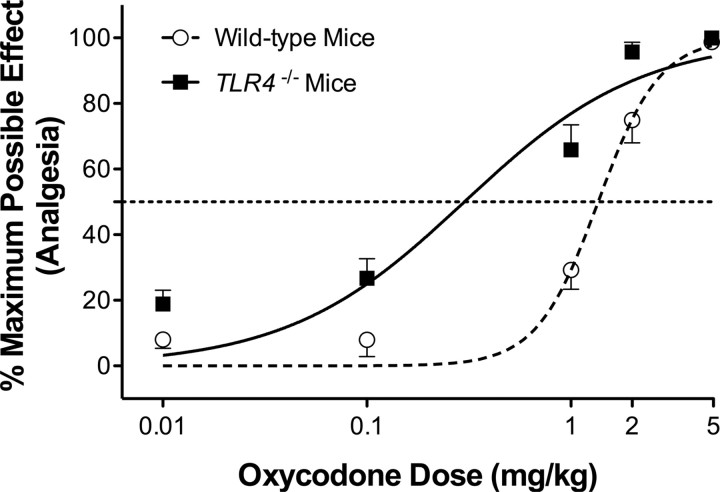
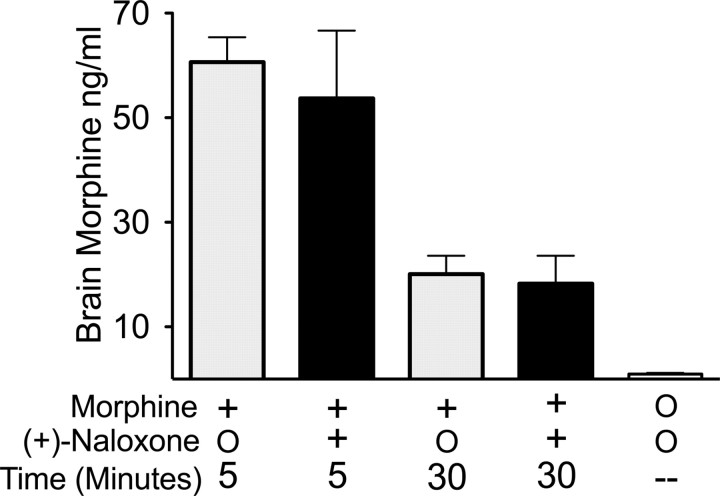
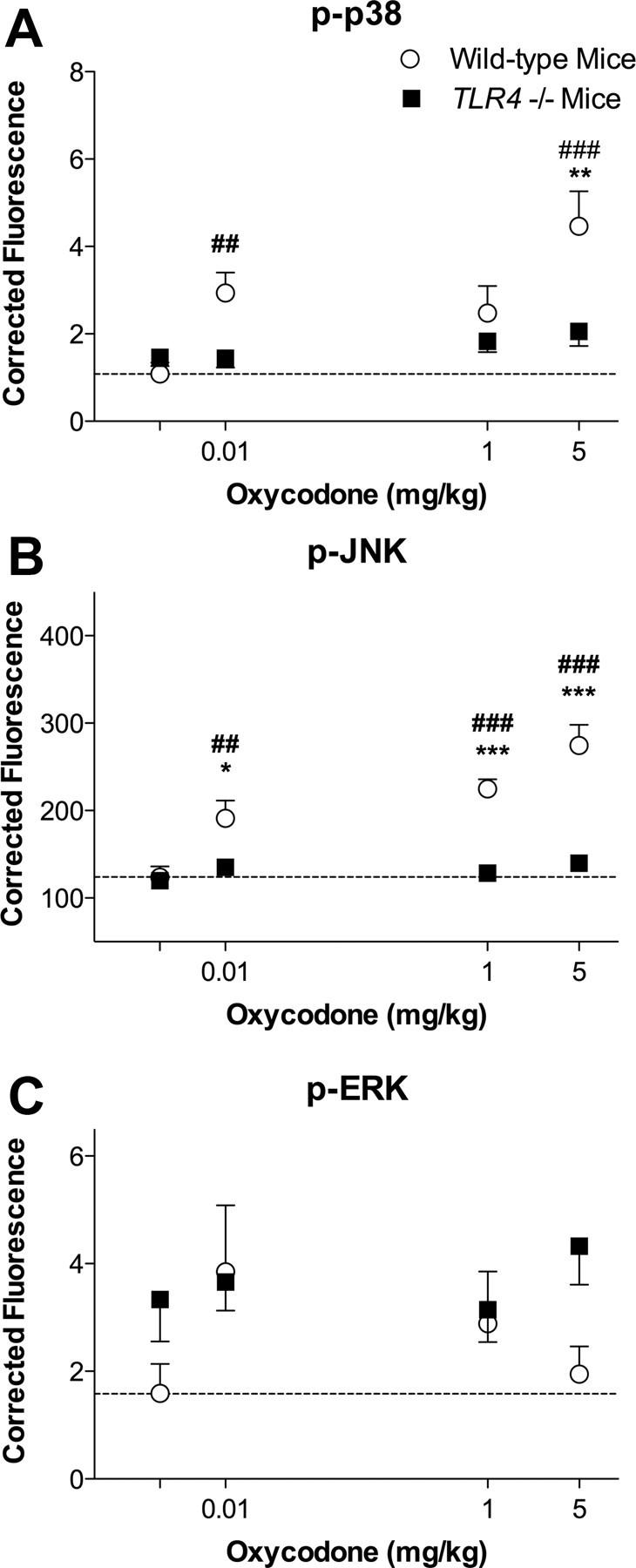
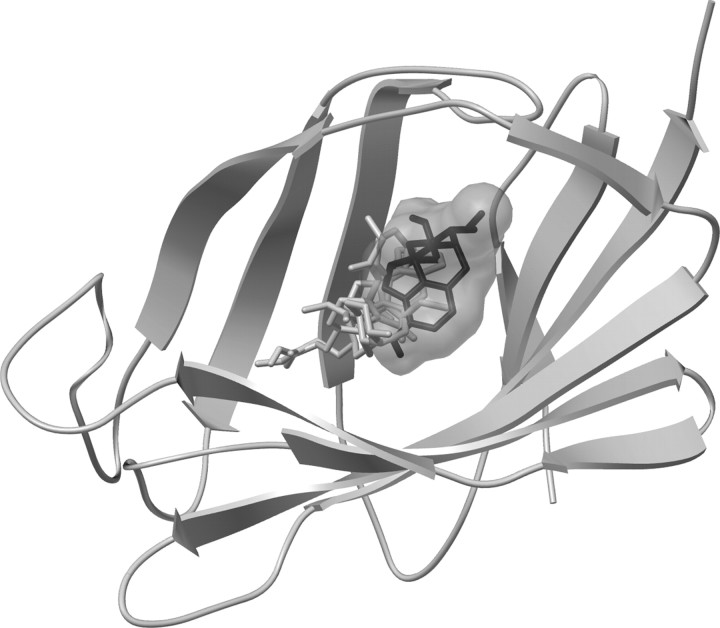
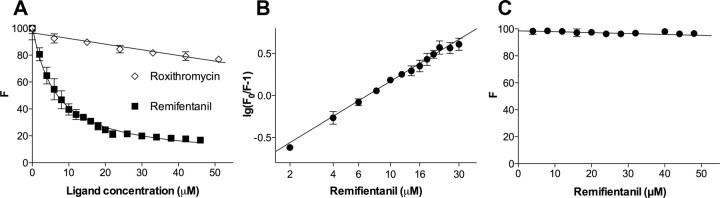
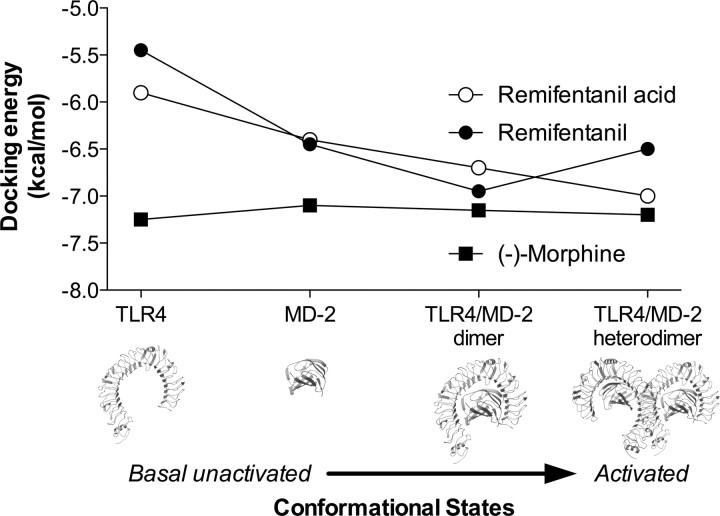
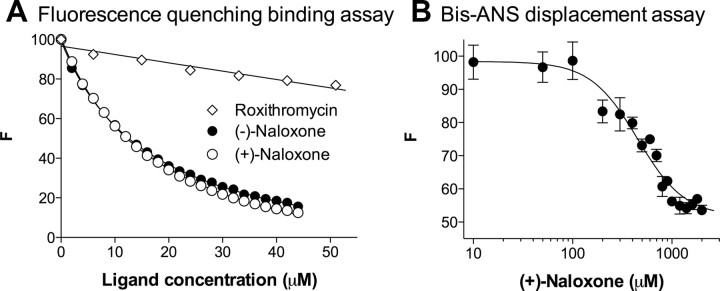
Opioid action was thought to exert reinforcing effects solely via the initial agonism of opioid receptors. Here, we present evidence for an additional novel contributor to opioid reward: the innate immune pattern-recognition receptor, toll-like receptor 4 (TLR4), and its MyD88-dependent signaling. Blockade of TLR4/MD2 by administration of the nonopioid, unnatural isomer of naloxone, (+)-naloxone (rats), or two independent genetic knock-outs of MyD88-TLR4-dependent signaling (mice), suppressed opioid-induced conditioned place preference. (+)-Naloxone also reduced opioid (remifentanil) self-administration (rats), another commonly used behavioral measure of drug reward. Moreover, pharmacological blockade of morphine-TLR4/MD2 activity potently reduced morphine-induced elevations of extracellular dopamine in rat nucleus accumbens, a region critical for opioid reinforcement. Importantly, opioid-TLR4 actions are not a unidirectional influence on opioid pharmacodynamics, since TLR4(-/-) mice had reduced oxycodone-induced p38 and JNK phosphorylation, while displaying potentiated analgesia. Similar to our recent reports of morphine-TLR4/MD2 binding, here we provide a combination of in silico and biophysical data to support (+)-naloxone and remifentanil binding to TLR4/MD2. Collectively, these data indicate that the actions of opioids at classical opioid receptors, together with their newly identified TLR4/MD2 actions, affect the mesolimbic dopamine system that amplifies opioid-induced elevations in extracellular dopamine levels, therefore possibly explaining altered opioid reward behaviors. Thus, the discovery of TLR4/MD2 recognition of opioids as foreign xenobiotic substances adds to the existing hypothesized neuronal reinforcement mechanisms, identifies a new drug target in TLR4/MD2 for the treatment of addictions, and provides further evidence supporting a role for central proinflammatory immune signaling in drug reward.
Figures
References
-
- Bowers MS, Kalivas PW. Forebrain astroglial plasticity is induced following withdrawal from repeated cocaine administration. Eur J Neurosci. 2003;17:1273–1278. - PubMed
-
- Bsibsi M, Ravid R, Gveric D, van Noort JM. Broad expression of Toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol. 2002;61:1013–1021. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- DA024044/DA/NIDA NIH HHS/United States
- R21 DA026950/DA/NIDA NIH HHS/United States
- NS067425/NS/NINDS NIH HHS/United States
- R03 DA025740/DA/NIDA NIH HHS/United States
- DA026950/DA/NIDA NIH HHS/United States
- ImNIH/Intramural NIH HHS/United States
- DA025740/DA/NIDA NIH HHS/United States
- R01 DE017782/DE/NIDCR NIH HHS/United States
- R01 DA023132/DA/NIDA NIH HHS/United States
- K05 DA024044/DA/NIDA NIH HHS/United States
- R03 DA027977/DA/NIDA NIH HHS/United States
- DA023132/DA/NIDA NIH HHS/United States
- DA027977/DA/NIDA NIH HHS/United States
- DE017782/DE/NIDCR NIH HHS/United States
- R21 NS067425/NS/NINDS NIH HHS/United States
- R01 DE021966/DE/NIDCR NIH HHS/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials