

TMEM106B, the risk gene for frontotemporal dementia, is regulated by the microRNA-132/212 cluster and affects progranulin pathways
- PMID: 22895706
- PMCID: PMC3446826
- DOI: 10.1523/JNEUROSCI.0521-12.2012
TMEM106B, the risk gene for frontotemporal dementia, is regulated by the microRNA-132/212 cluster and affects progranulin pathways
Abstract
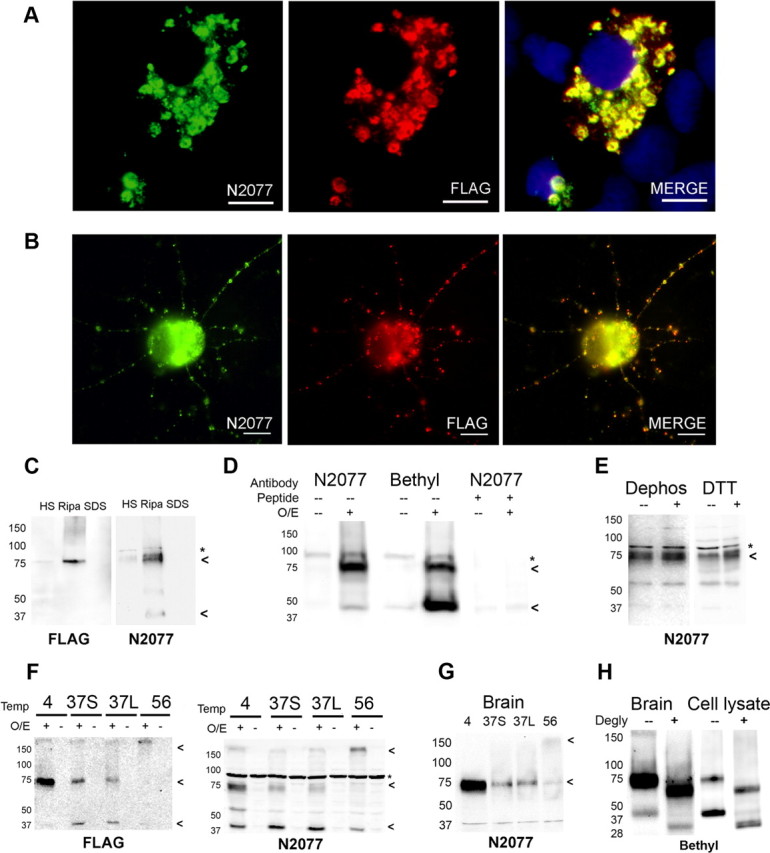
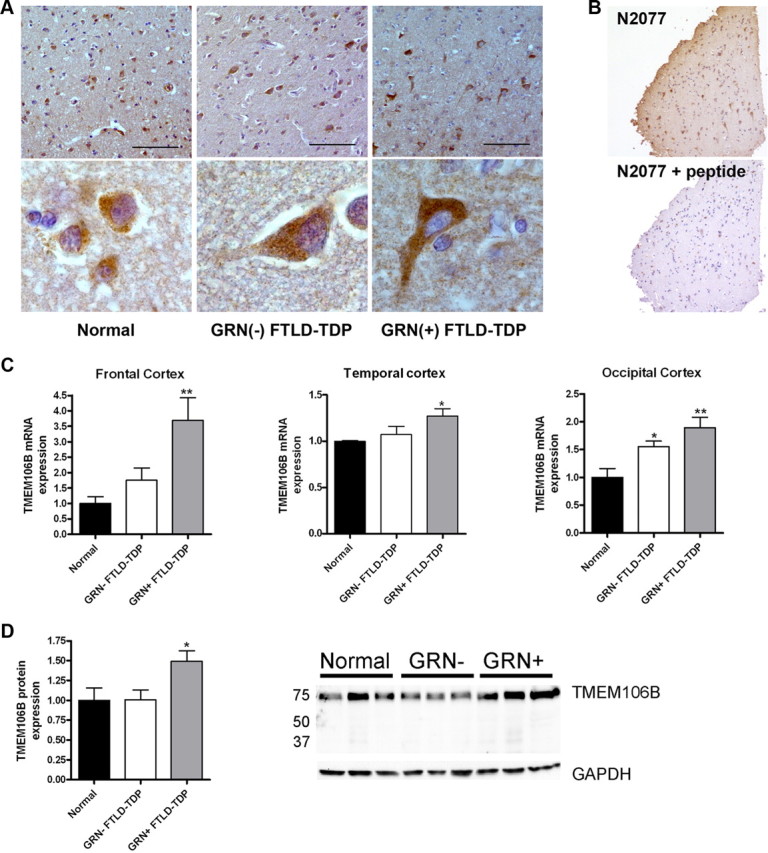
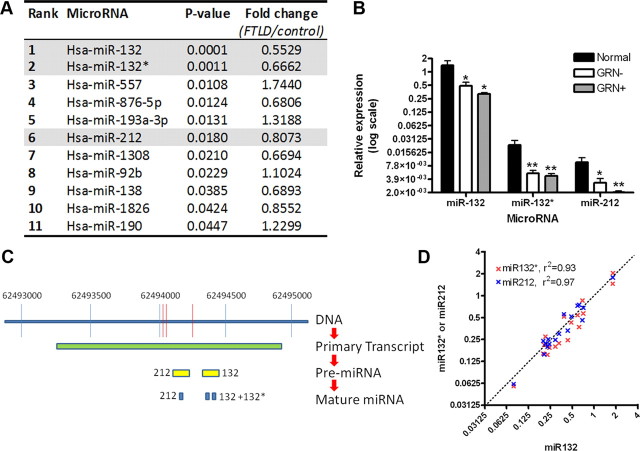
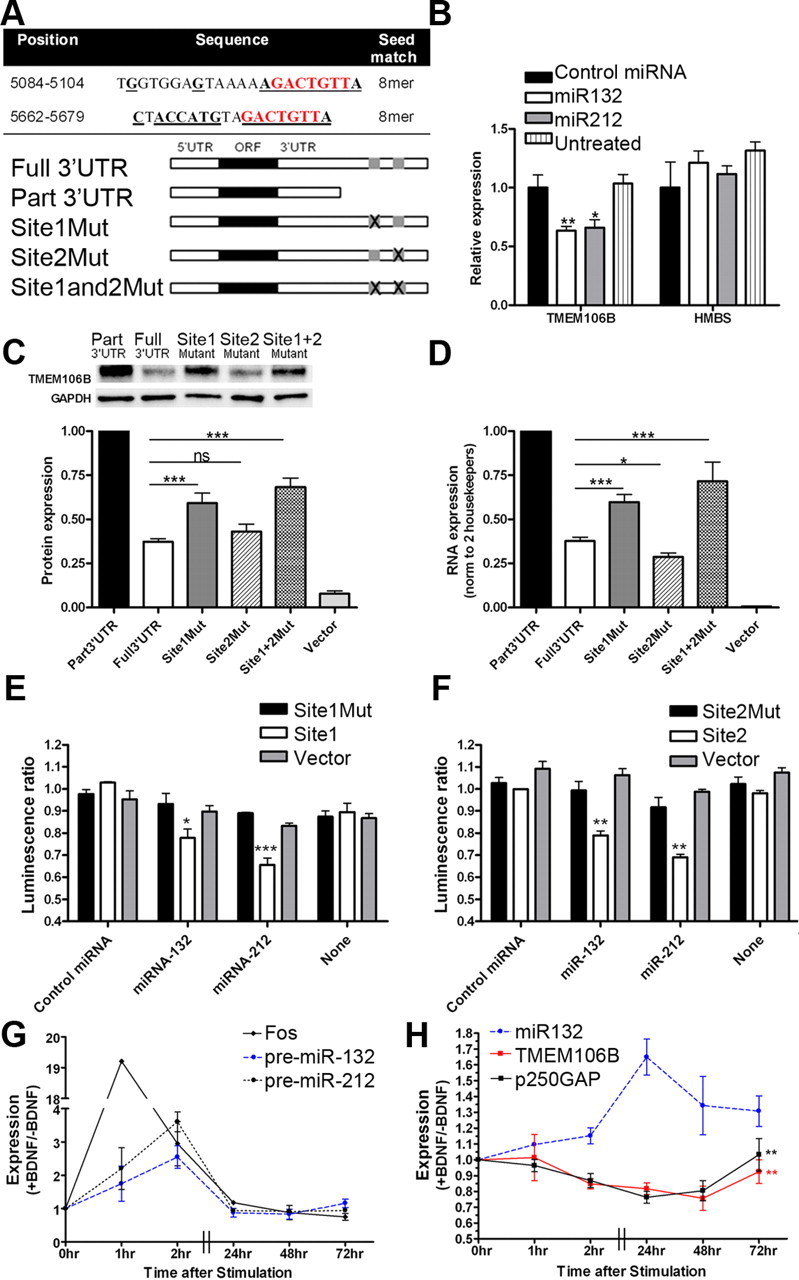
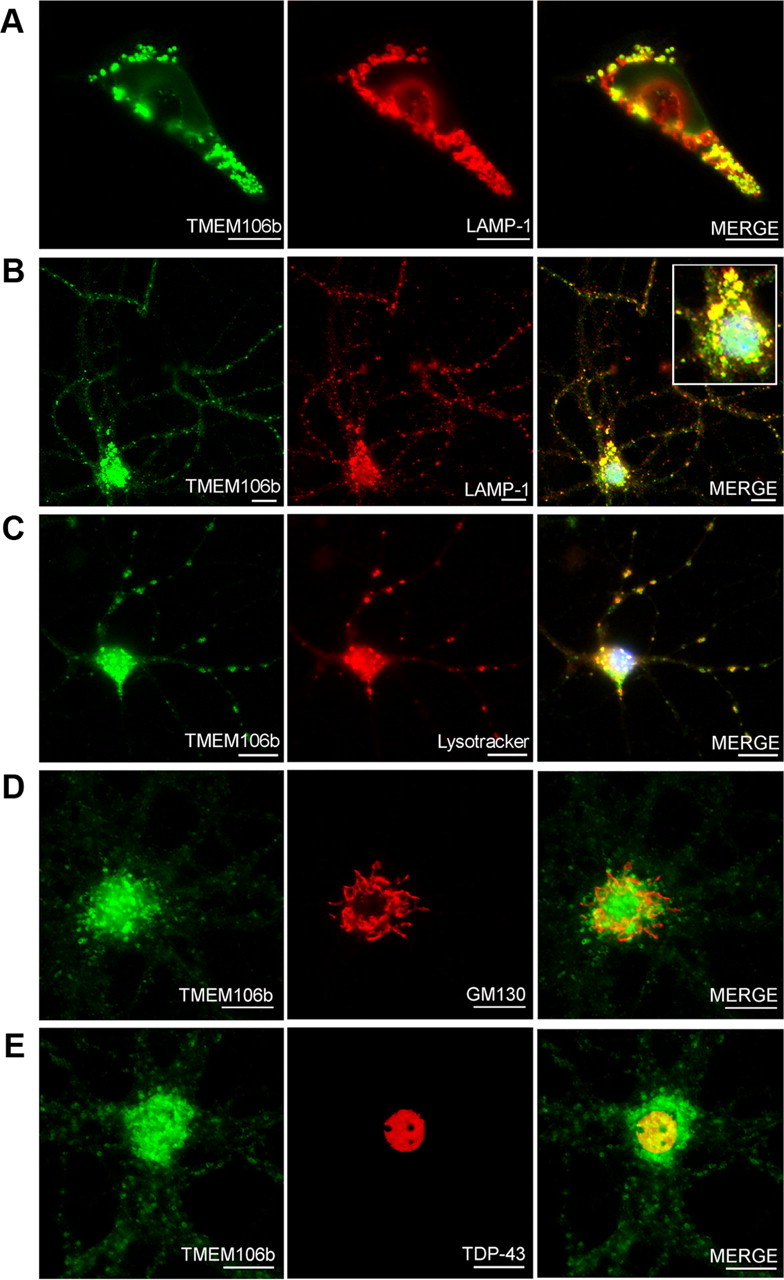
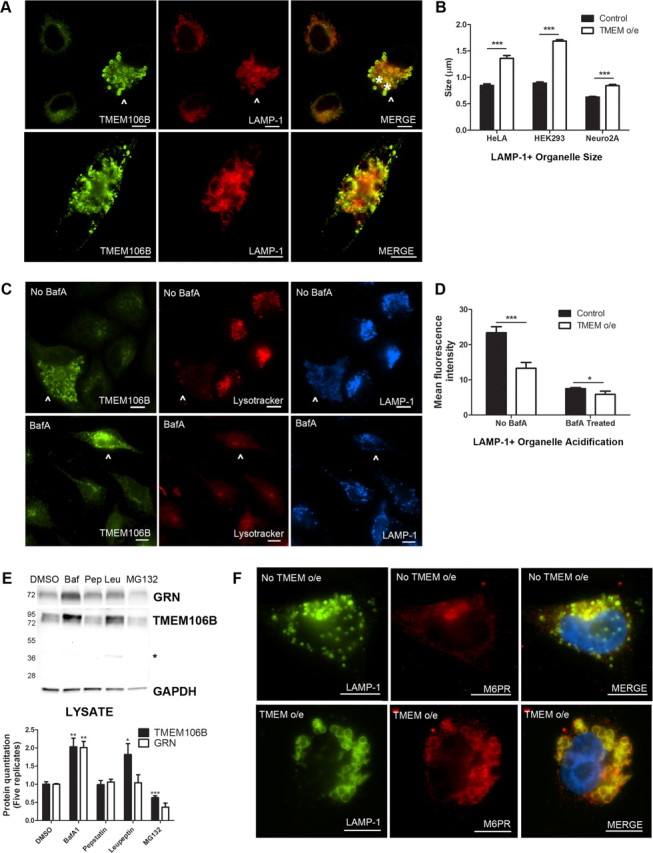
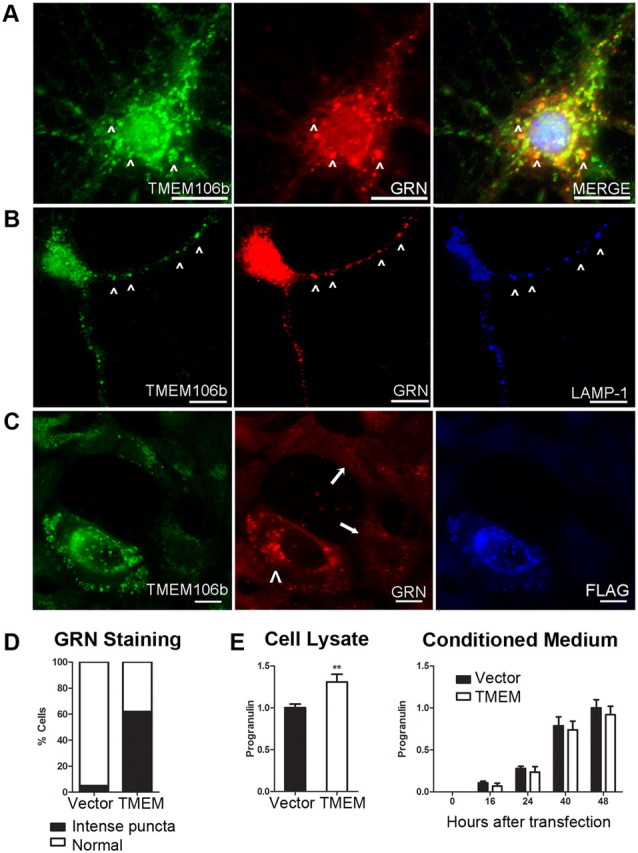
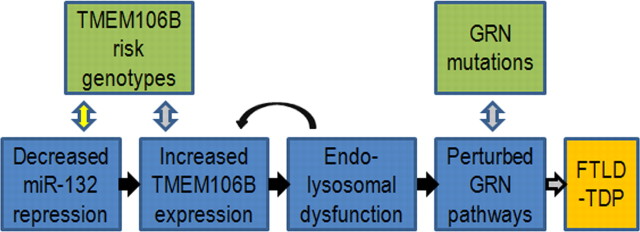
Frontotemporal lobar degeneration with TDP-43 inclusions (FTLD-TDP) is a fatal neurodegenerative disease with no available treatments. Mutations in the progranulin gene (GRN) causing impaired production or secretion of progranulin are a common Mendelian cause of FTLD-TDP; additionally, common variants at chromosome 7p21 in the uncharacterized gene TMEM106B were recently linked by genome-wide association to FTLD-TDP with and without GRN mutations. Here we show that TMEM106B is neuronally expressed in postmortem human brain tissue, and that expression levels are increased in FTLD-TDP brain. Furthermore, using an unbiased, microarray-based screen of >800 microRNAs (miRs), we identify microRNA-132 as the top microRNA differentiating FTLD-TDP and control brains, with <50% normal expression levels of three members of the microRNA-132 cluster (microRNA-132, microRNA-132*, and microRNA-212) in disease. Computational analyses, corroborated empirically, demonstrate that the top mRNA target of both microRNA-132 and microRNA-212 is TMEM106B; both microRNAs repress TMEM106B expression through shared microRNA-132/212 binding sites in the TMEM106B 3'UTR. Increasing TMEM106B expression to model disease results in enlargement and poor acidification of endo-lysosomes, as well as impairment of mannose-6-phosphate-receptor trafficking. Finally, endogenous neuronal TMEM106B colocalizes with progranulin in late endo-lysosomes, and TMEM106B overexpression increases intracellular levels of progranulin. Thus, TMEM106B is an FTLD-TDP risk gene, with microRNA-132/212 depression as an event which can lead to aberrant overexpression of TMEM106B, which in turn alters progranulin pathways. Evidence for this pathogenic cascade includes the striking convergence of two independent, genomic-scale screens on a microRNA:mRNA regulatory pair. Our findings open novel directions for elucidating miR-based therapies in FTLD-TDP.
Figures
References
-
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–611. - PubMed
-
- Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. - PubMed
-
- Benajiba L, Le Ber I, Camuzat A, Lacoste M, Thomas-Anterion C, Couratier P, Legallic S, Salachas F, Hannequin D, Decousus M, Lacomblez L, Guedj E, Golfier V, Camu W, Dubois B, Campion D, Meininger V, Brice A. TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann Neurol. 2009;65:470–473. - PubMed
-
- Capell A, Liebscher S, Fellerer K, Brouwers N, Willem M, Lammich S, Gijselinck I, Bittner T, Carlson AM, Sasse F, Kunze B, Steinmetz H, Jansen R, Dormann D, Sleegers K, Cruts M, Herms J, Van Broeckhoven C, Haass C. Rescue of progranulin deficiency associated with frontotemporal lobar degeneration by alkalizing reagents and inhibition of vacuolar ATPase. J Neurosci. 2011;31:1885–1894. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous