

The cytoplasmic tail domain of influenza B virus hemagglutinin is important for its incorporation into virions but is not essential for virus replication in cell culture in the presence of compensatory mutations
- PMID: 22896616
- PMCID: PMC3486280
- DOI: 10.1128/JVI.01479-12
The cytoplasmic tail domain of influenza B virus hemagglutinin is important for its incorporation into virions but is not essential for virus replication in cell culture in the presence of compensatory mutations
Abstract
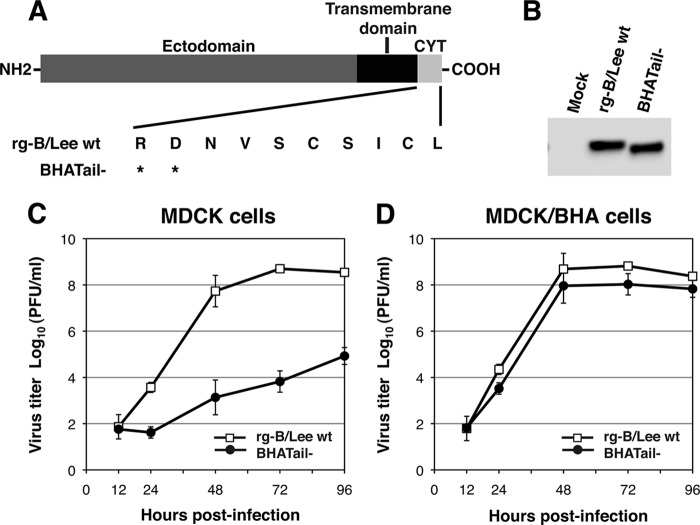
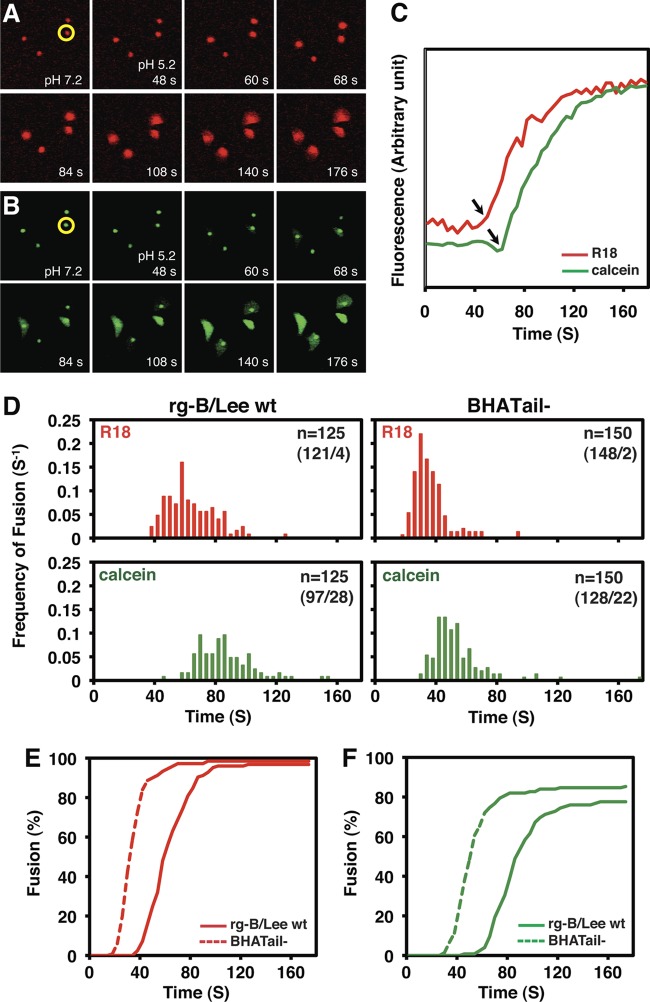
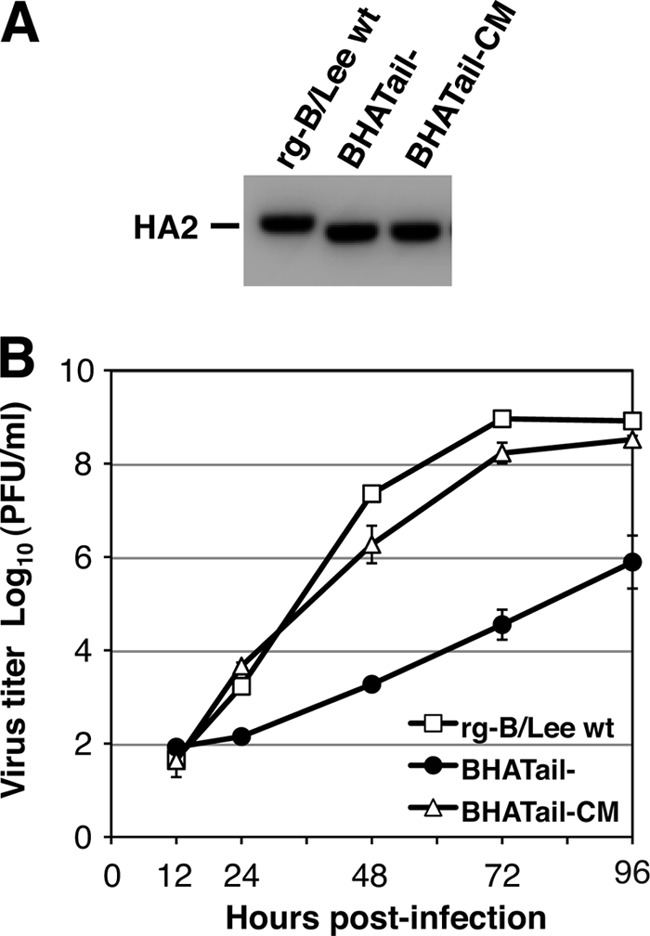
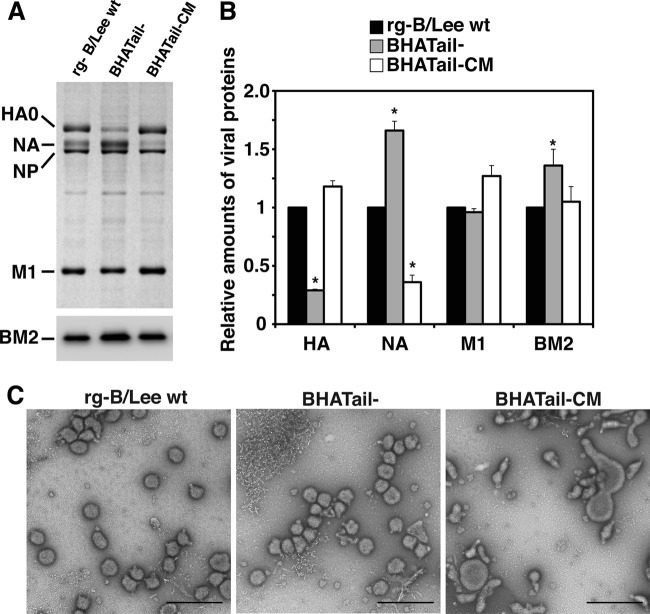
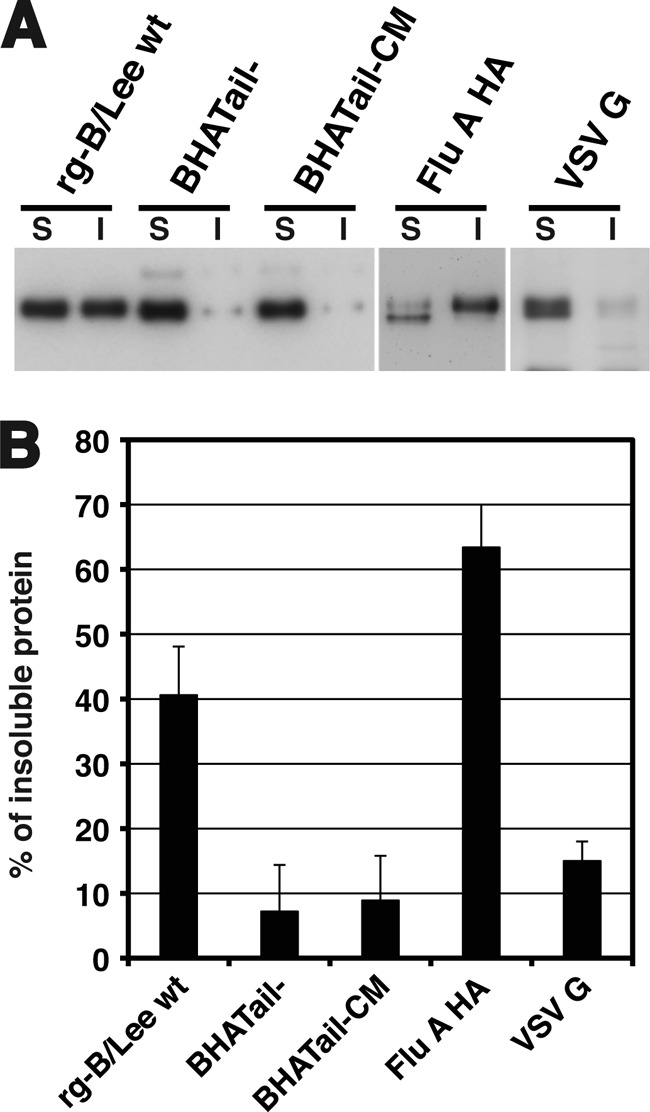
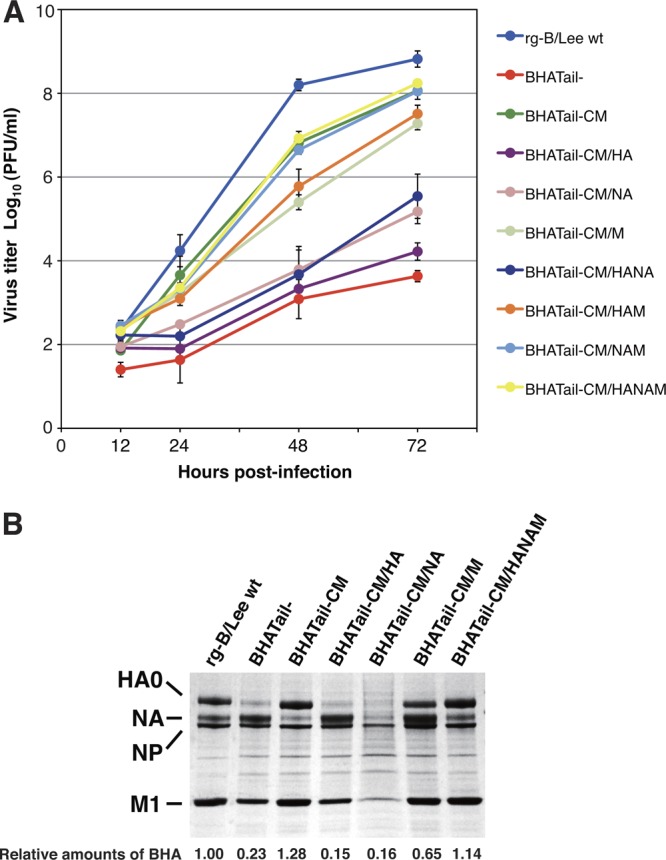
Influenza B virus hemagglutinin (BHA) contains a predicted cytoplasmic tail of 10 amino acids that are highly conserved among influenza B viruses. To understand the role of this cytoplasmic tail in infectious virus production, we used reverse genetics to generate a recombinant influenza B virus lacking the BHA cytoplasmic tail domain. The resulting virus, designated BHATail(-), had a titer approximately 5 log units lower than that of wild-type virus but grew normally when BHA was supplemented in trans by BHA-expressing cells. Although the levels of BHA cell surface expression were indistinguishable between truncated and wild-type BHA, the BHATail(-) virus produced particles containing dramatically less BHA. Moreover, removal of the cytoplasmic tail abrogated the association of BHA with Triton X-100-insoluble lipid rafts. Interestingly, long-term culture of a virus lacking the BHA cytoplasmic tail in Madin-Darby canine kidney (MDCK) cells yielded a mutant with infectivities somewhat similar to that of wild-type virus. Sequencing revealed that the mutant virus retained the original cytoplasmic tail deletion but acquired additional mutations in its BHA, neuraminidase (NA), and M1 proteins. Viral growth kinetic analysis showed that replication of BHA cytoplasmic tailless viruses could be improved by compensatory mutations in the NA and M1 proteins. These findings indicate that the cytoplasmic tail domain of BHA is important for efficient incorporation of BHA into virions and tight lipid raft association. They also demonstrate that the domain is not absolutely required for virus viability in cell culture in the presence of compensatory mutations.
Figures

Similar articles
-
Influenza A virus hemagglutinin and neuraminidase mutually accelerate their apical targeting through clustering of lipid rafts.J Virol. 2014 Sep 1;88(17):10039-55. doi: 10.1128/JVI.00586-14. Epub 2014 Jun 25. J Virol. 2014. PMID: 24965459 Free PMC article.
-
The cytoplasmic tail of influenza A virus neuraminidase (NA) affects NA incorporation into virions, virion morphology, and virulence in mice but is not essential for virus replication.J Virol. 1996 Feb;70(2):873-9. doi: 10.1128/JVI.70.2.873-879.1996. J Virol. 1996. PMID: 8551626 Free PMC article.
-
Cytoplasmic domain of influenza B virus BM2 protein plays critical roles in production of infectious virus.J Virol. 2008 Jan;82(2):728-39. doi: 10.1128/JVI.01752-07. Epub 2007 Nov 7. J Virol. 2008. PMID: 17989175 Free PMC article.
-
Transport of viral proteins to the apical membranes and interaction of matrix protein with glycoproteins in the assembly of influenza viruses.Virus Res. 2001 Sep;77(1):61-9. doi: 10.1016/s0168-1702(01)00266-0. Virus Res. 2001. PMID: 11451488 Review.
-
Functional balance between neuraminidase and haemagglutinin in influenza viruses.Clin Microbiol Infect. 2016 Dec;22(12):975-983. doi: 10.1016/j.cmi.2016.07.007. Epub 2016 Jul 15. Clin Microbiol Infect. 2016. PMID: 27424943 Review.
Cited by
-
Identification of a Novel Universal Potential Epitope on the Cytoplasmic Tail of H7N9 Virus Hemagglutinin.Virol Sin. 2019 Jun;34(3):334-337. doi: 10.1007/s12250-019-00110-7. Epub 2019 Apr 23. Virol Sin. 2019. PMID: 31016481 Free PMC article. No abstract available.
-
The Influenza B Virus Hemagglutinin Head Domain Is Less Tolerant to Transposon Mutagenesis than That of the Influenza A Virus.J Virol. 2018 Jul 31;92(16):e00754-18. doi: 10.1128/JVI.00754-18. Print 2018 Aug 15. J Virol. 2018. PMID: 29899093 Free PMC article.
-
Two Cytoplasmic Acylation Sites and an Adjacent Hydrophobic Residue, but No Other Conserved Amino Acids in the Cytoplasmic Tail of HA from Influenza A Virus Are Crucial for Virus Replication.Viruses. 2015 Dec 8;7(12):6458-75. doi: 10.3390/v7122950. Viruses. 2015. PMID: 26670246 Free PMC article.
References
-
- Baigent SJ, McCauley JW. 2001. Glycosylation of haemagglutinin and stalk-length of neuraminidase combine to regulate the growth of avian influenza viruses in tissue culture. Virus Res. 79:177–185 - PubMed
-
- Bourmakina SV, Garcia-Sastre A. 2003. Reverse genetics studies on the filamentous morphology of influenza A virus. J. Gen. Virol. 84:517–527 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
