

Redox control of cardiac excitability
- PMID: 22897788
- PMCID: PMC3526898
- DOI: 10.1089/ars.2011.4234
Redox control of cardiac excitability
Abstract
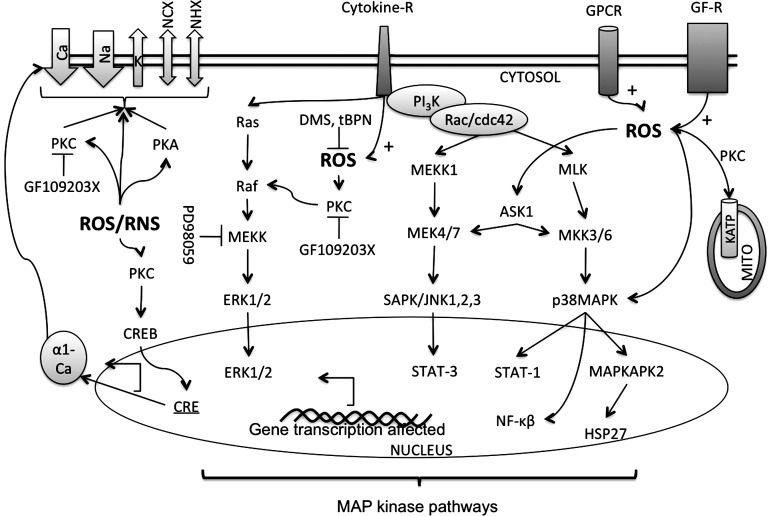
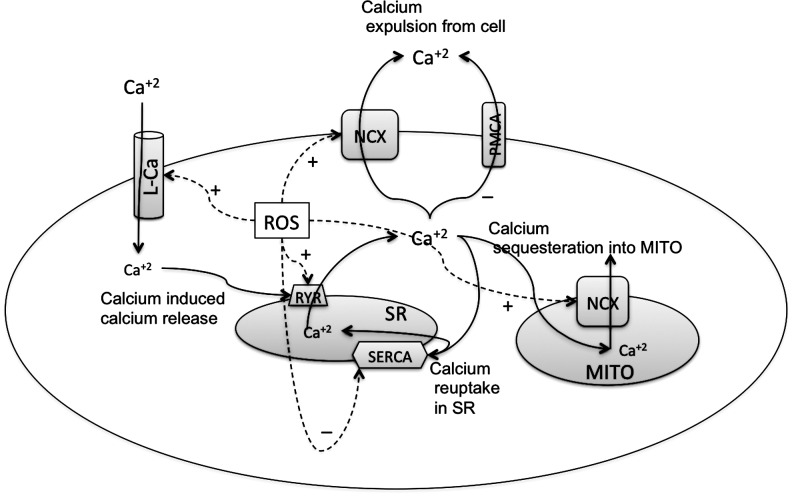
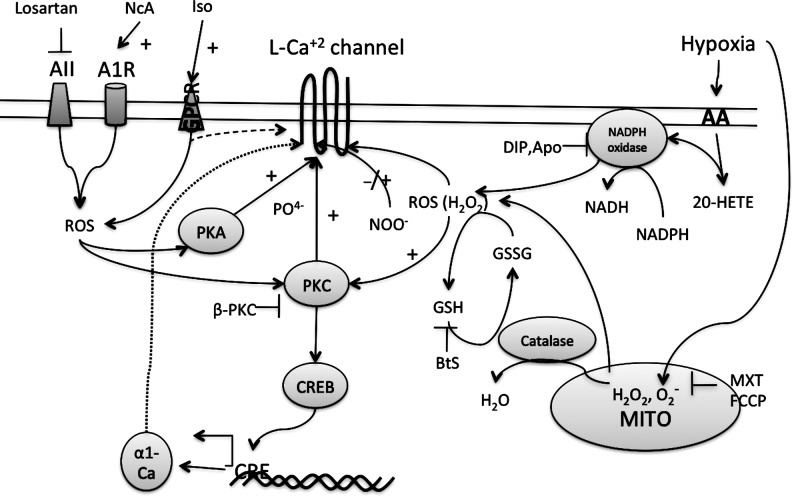
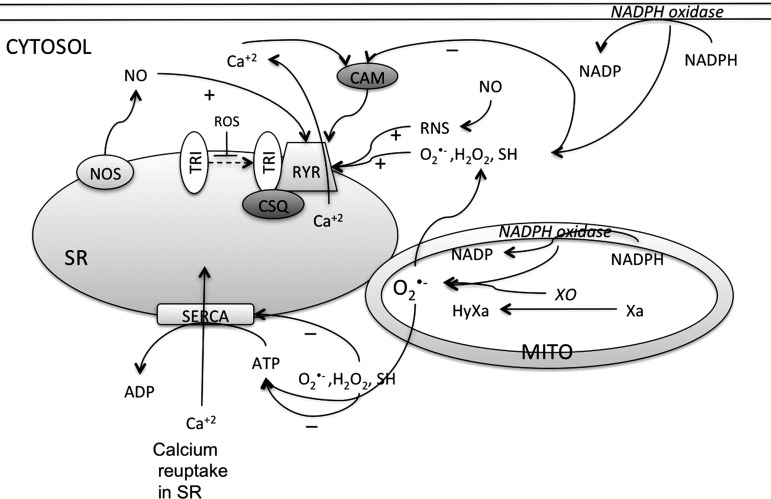
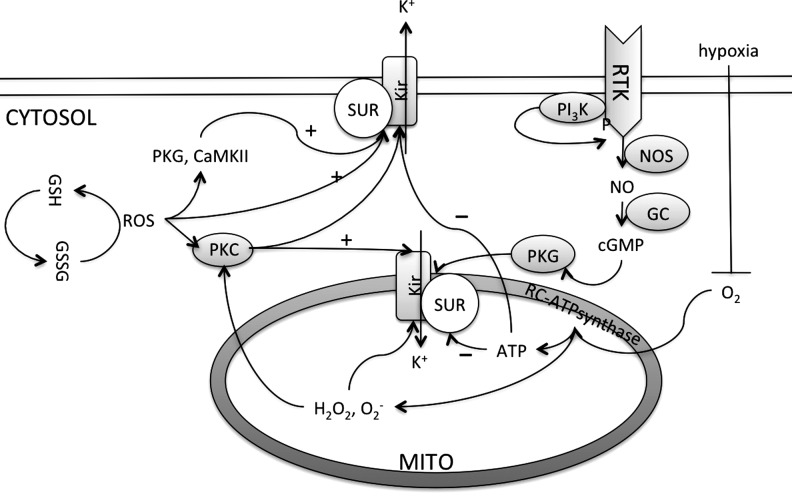
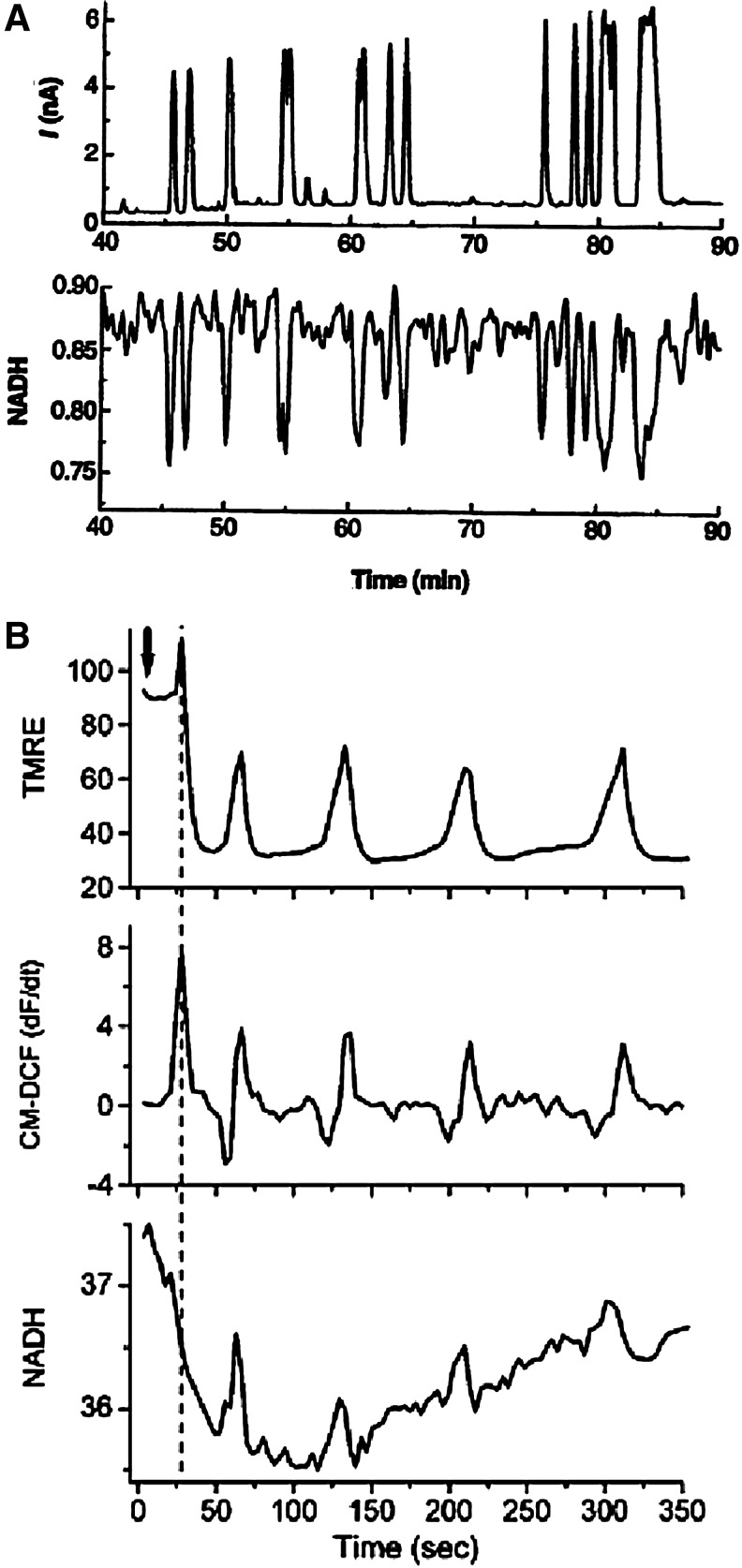
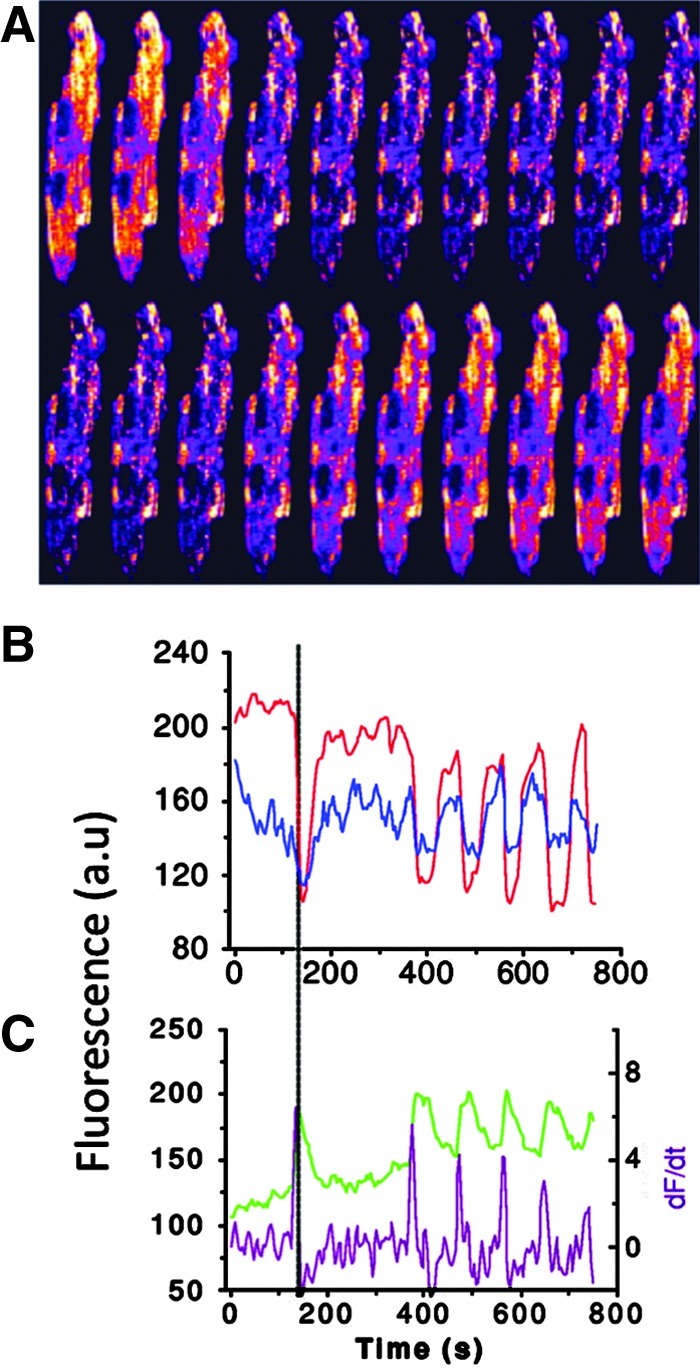
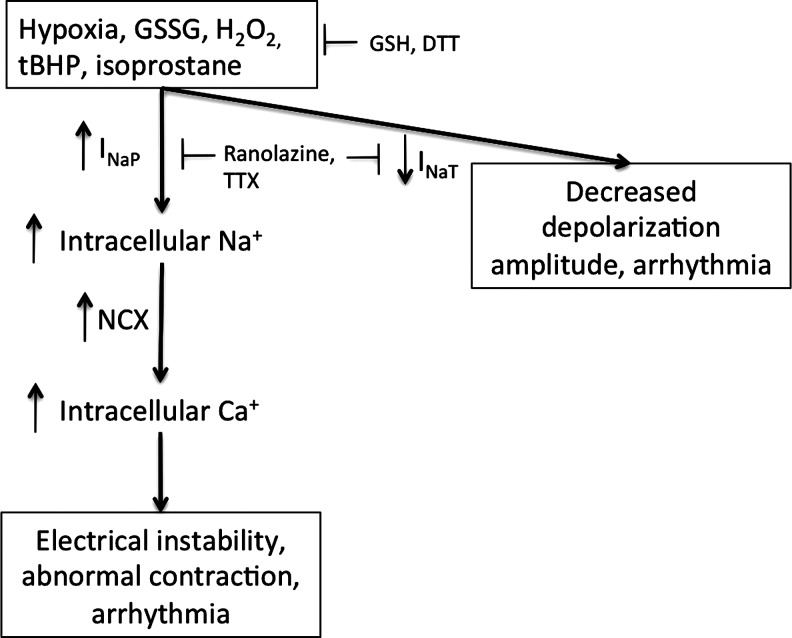
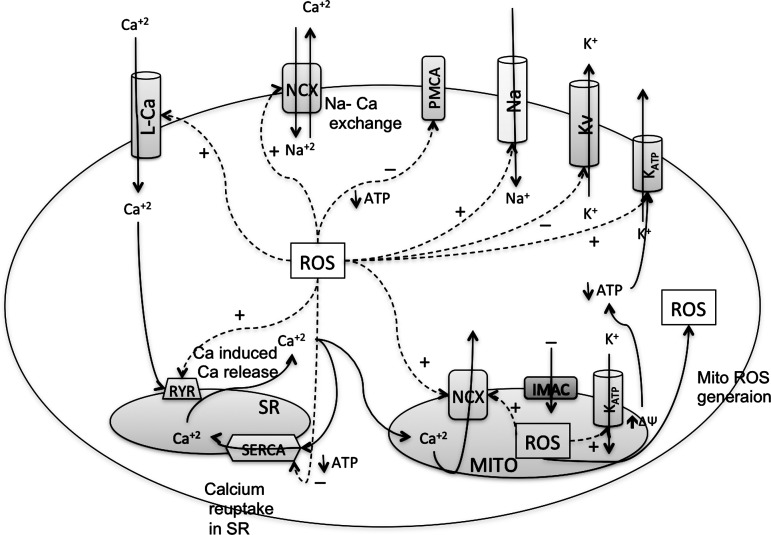
Reactive oxygen species (ROS) have been associated with various human diseases, and considerable attention has been paid to investigate their physiological effects. Various ROS are synthesized in the mitochondria and accumulate in the cytoplasm if the cellular antioxidant defense mechanism fails. The critical balance of this ROS synthesis and antioxidant defense systems is termed the redox system of the cell. Various cardiovascular diseases have also been affected by redox to different degrees. ROS have been indicated as both detrimental and protective, via different cellular pathways, for cardiac myocyte functions, electrophysiology, and pharmacology. Mostly, the ROS functions depend on the type and amount of ROS synthesized. While the literature clearly indicates ROS effects on cardiac contractility, their effects on cardiac excitability are relatively under appreciated. Cardiac excitability depends on the functions of various cardiac sarcolemal or mitochondrial ion channels carrying various depolarizing or repolarizing currents that also maintain cellular ionic homeostasis. ROS alter the functions of these ion channels to various degrees to determine excitability by affecting the cellular resting potential and the morphology of the cardiac action potential. Thus, redox balance regulates cardiac excitability, and under pathological regulation, may alter action potential propagation to cause arrhythmia. Understanding how redox affects cellular excitability may lead to potential prophylaxis or treatment for various arrhythmias. This review will focus on the studies of redox and cardiac excitation.
Figures
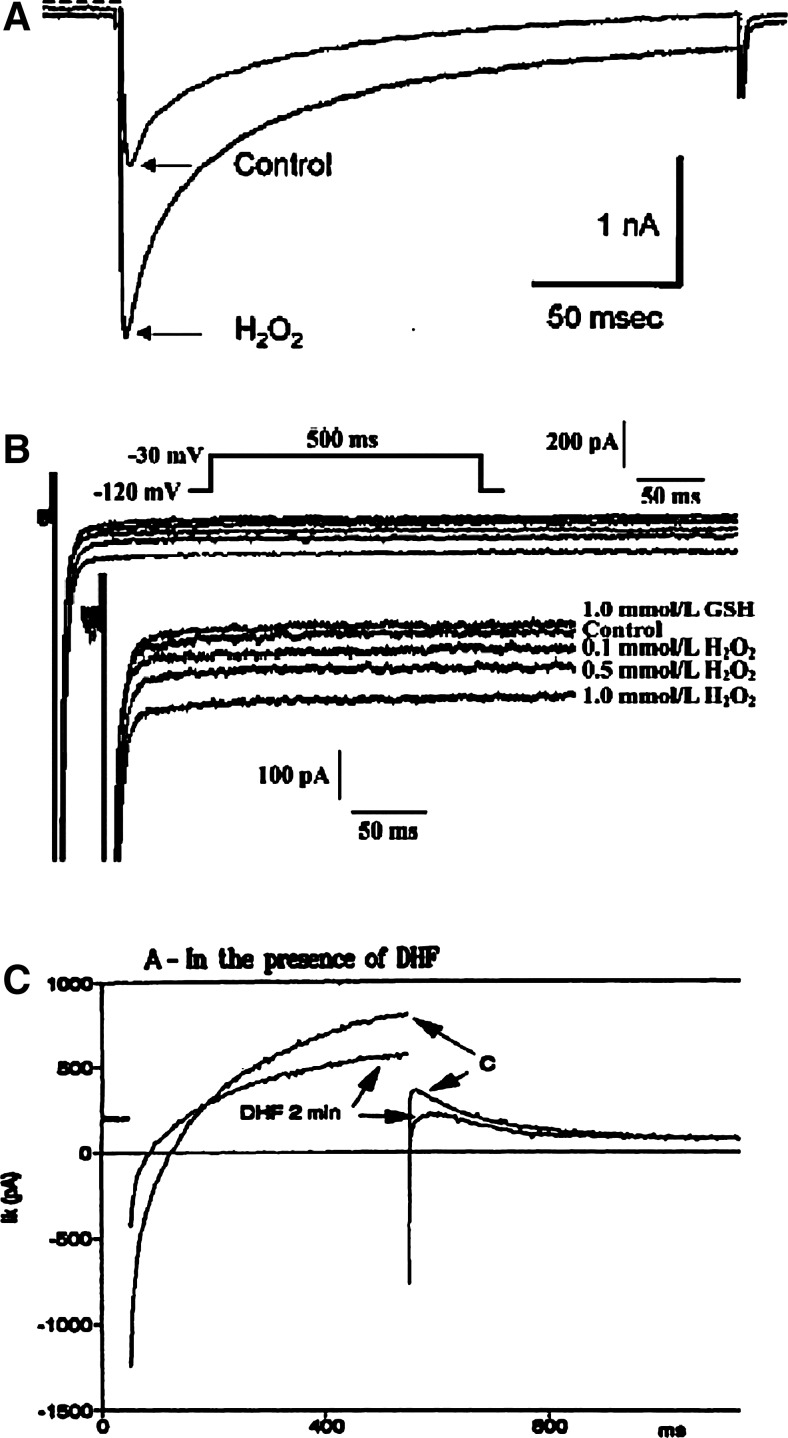
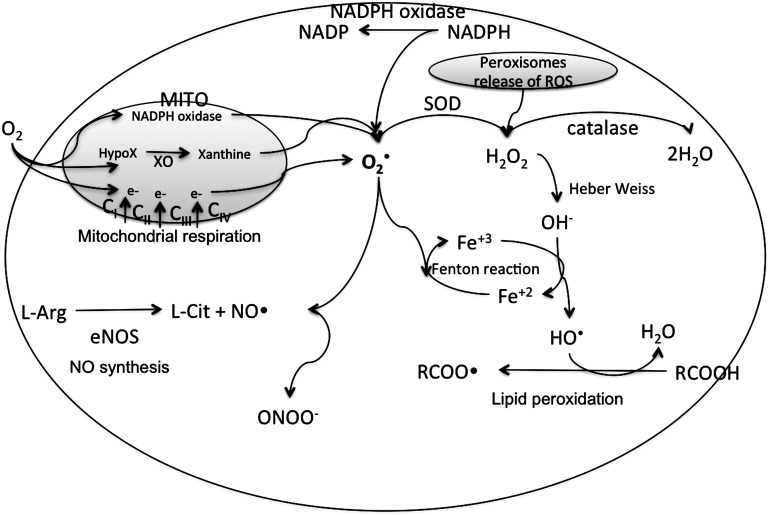
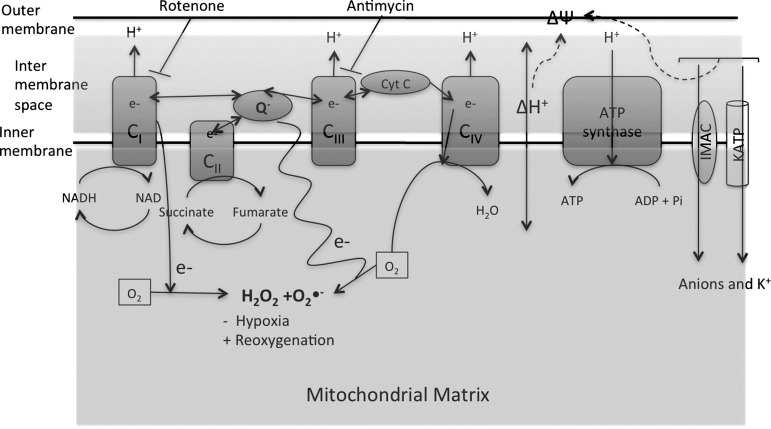
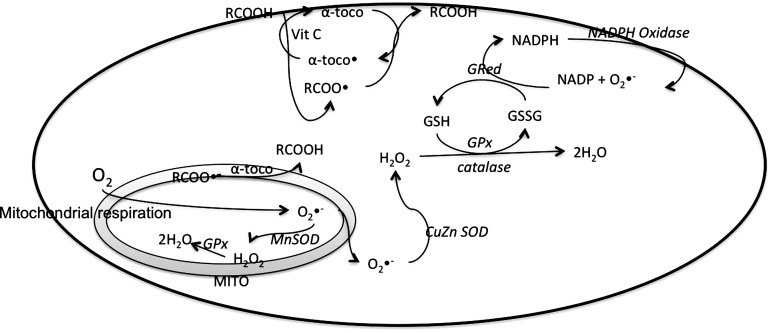
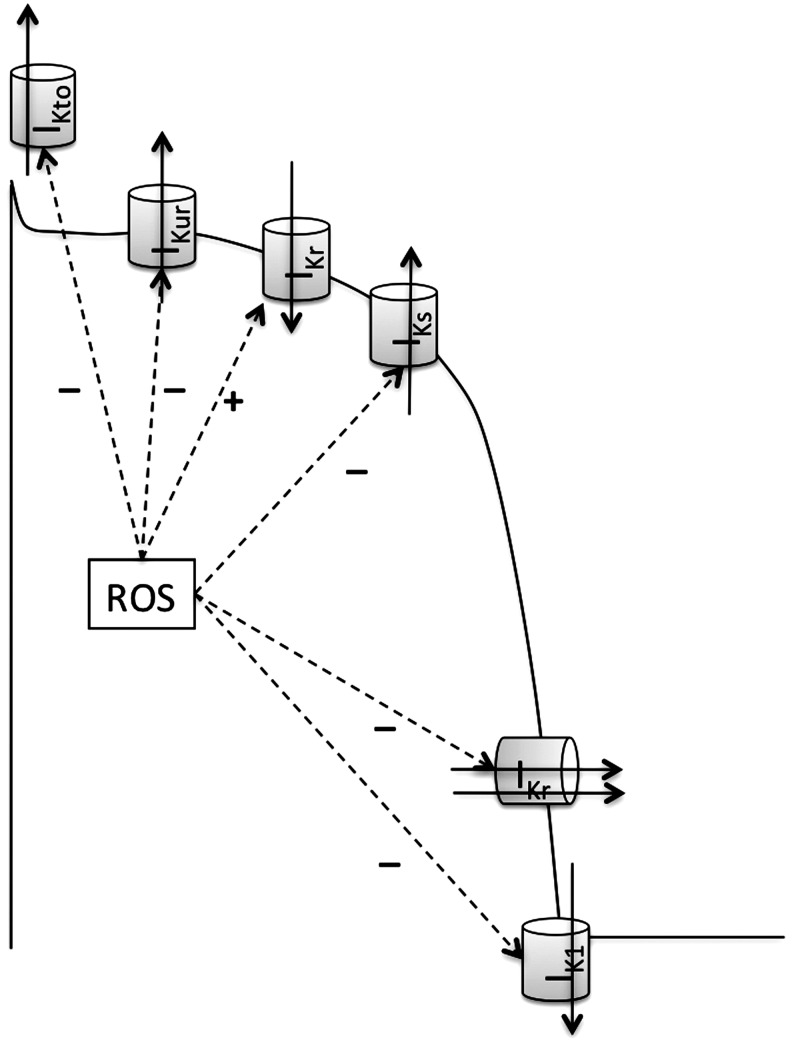
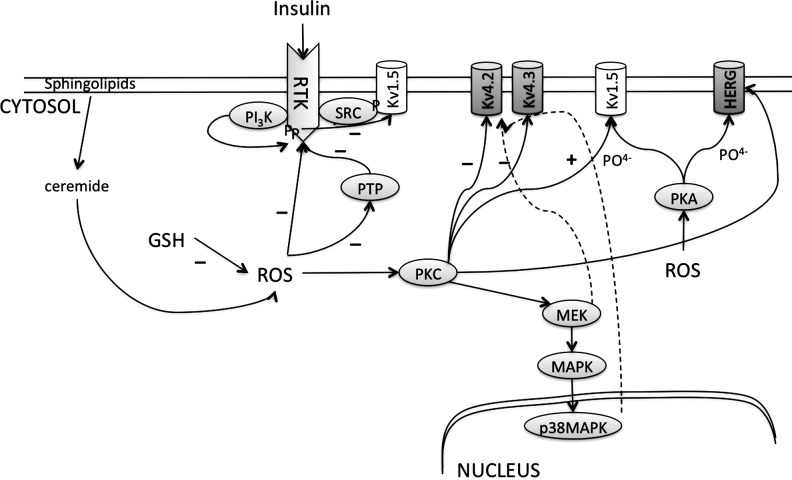
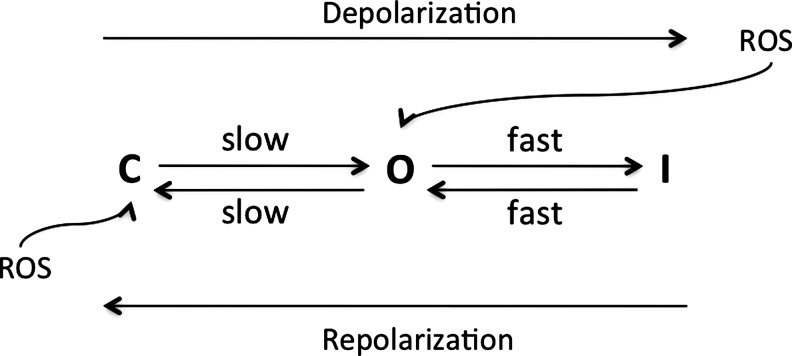
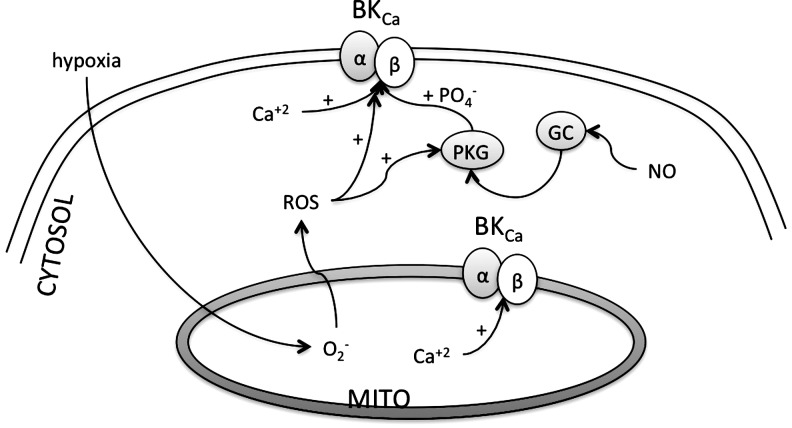
References
-
- Abriel H. Cardiac sodium channel Na(v)1.5 and interacting proteins: Physiology and pathophysiology. J Mol Cell Cardiol. 2010;48:2–11. - PubMed
-
- Adachi T. Weisbrod RM. Pimentel DR. Ying J. Sharov VS. Schoneich C. Cohen RA. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med. 2004;10:1200–1207. - PubMed
-
- Ahern GP. Hsu SF. Klyachko VA. Jackson MB. Induction of persistent sodium current by exogenous and endogenous nitric oxide. J Biol Chem. 2000;275:28810–28815. - PubMed
-
- Ahmmed GU. Xu Y. Hong Dong P. Zhang Z. Eiserich J. Chiamvimonvat N. Nitric oxide modulates cardiac Na(+) channel via protein kinase A and protein kinase G. Circ Res. 2001;89:1005–1013. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
