

ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression
- PMID: 22897849
- PMCID: PMC3422511
- DOI: 10.1016/j.ccr.2012.06.032
ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression
Abstract
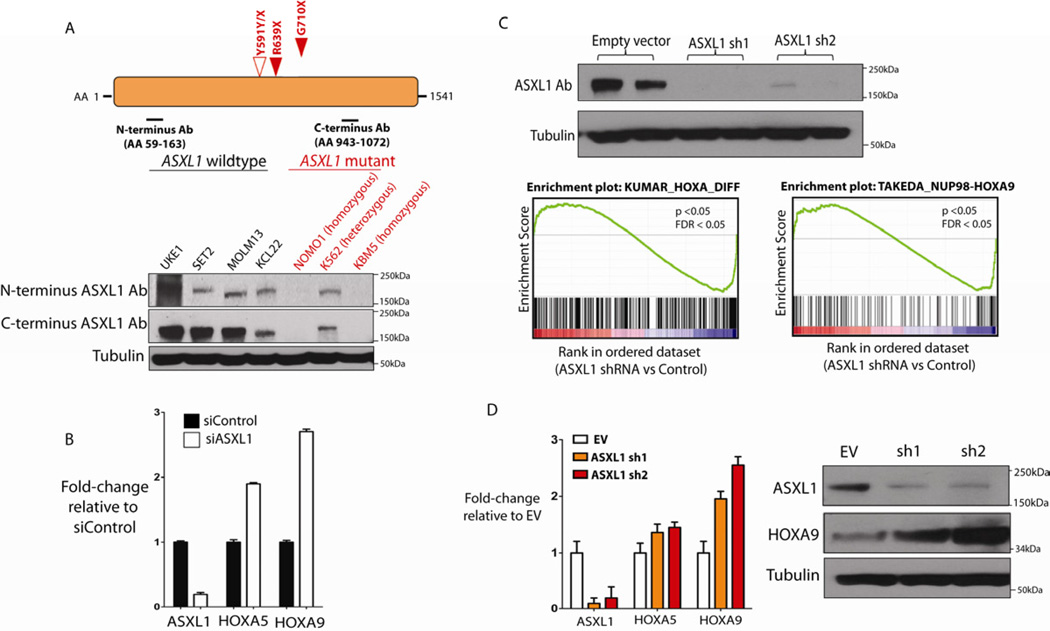
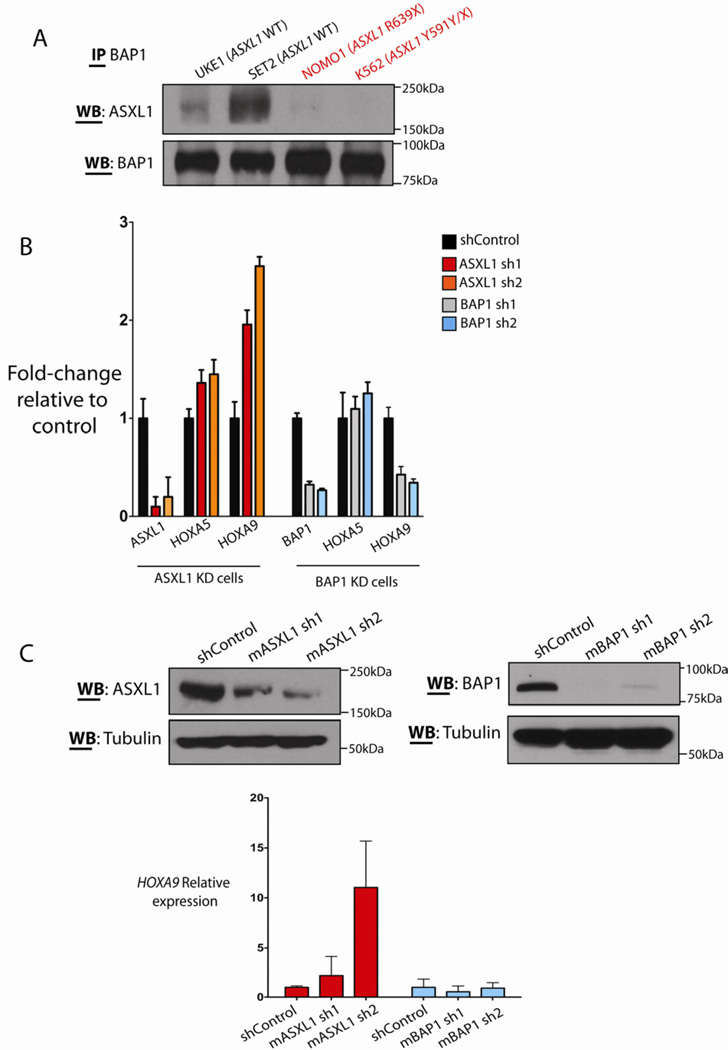
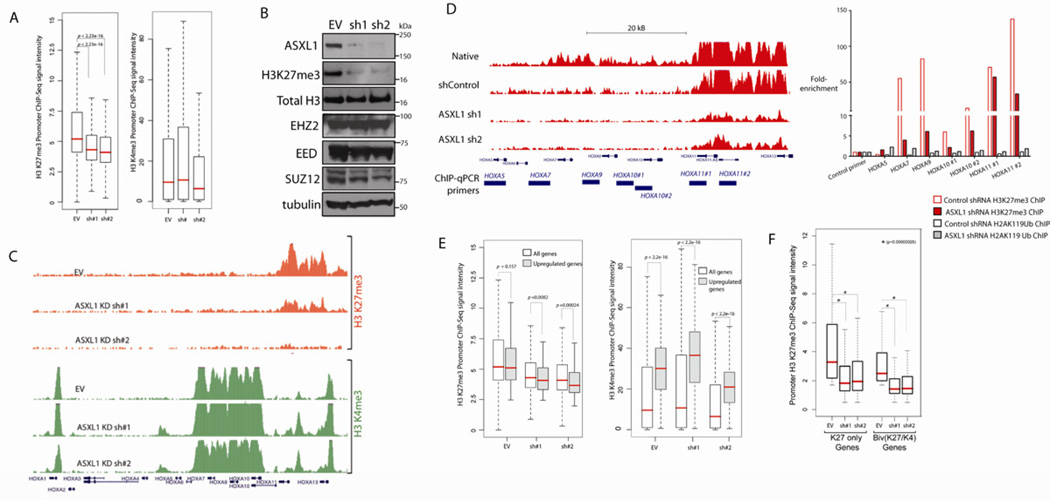
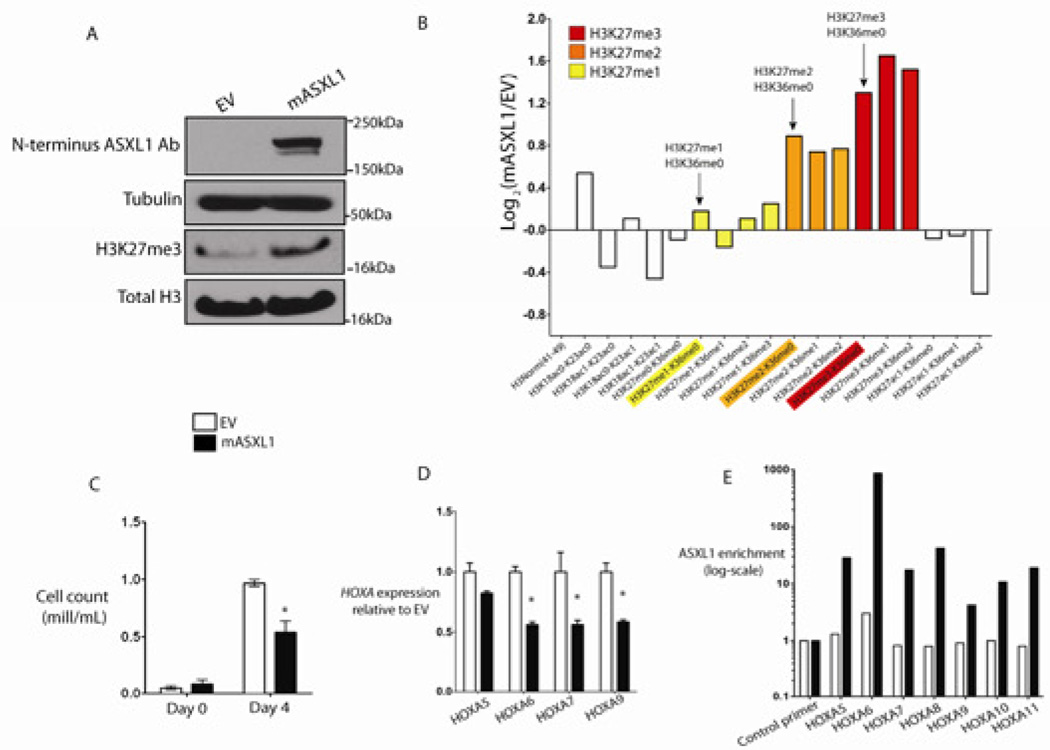
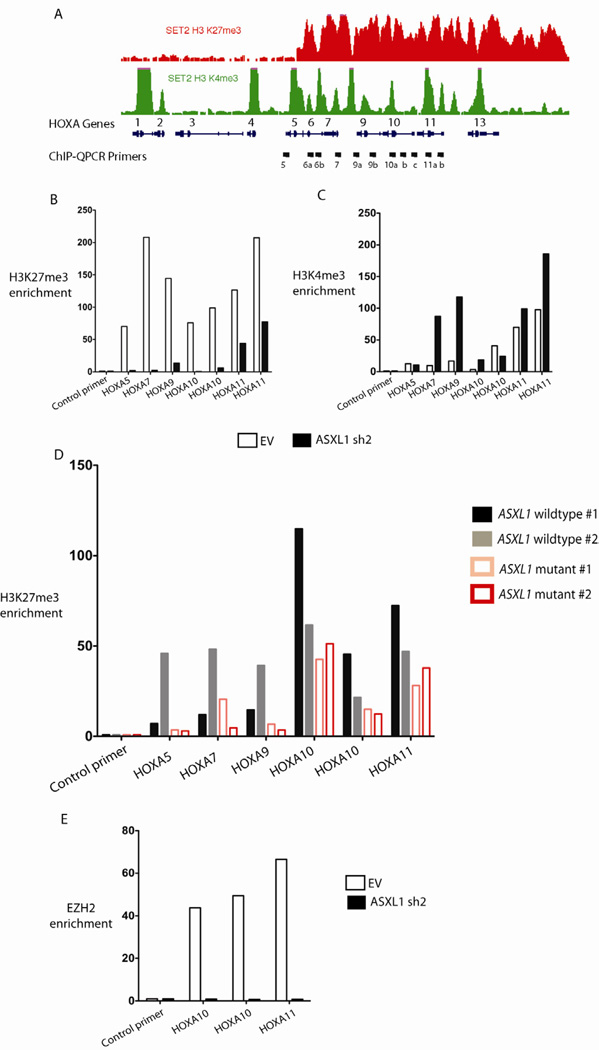
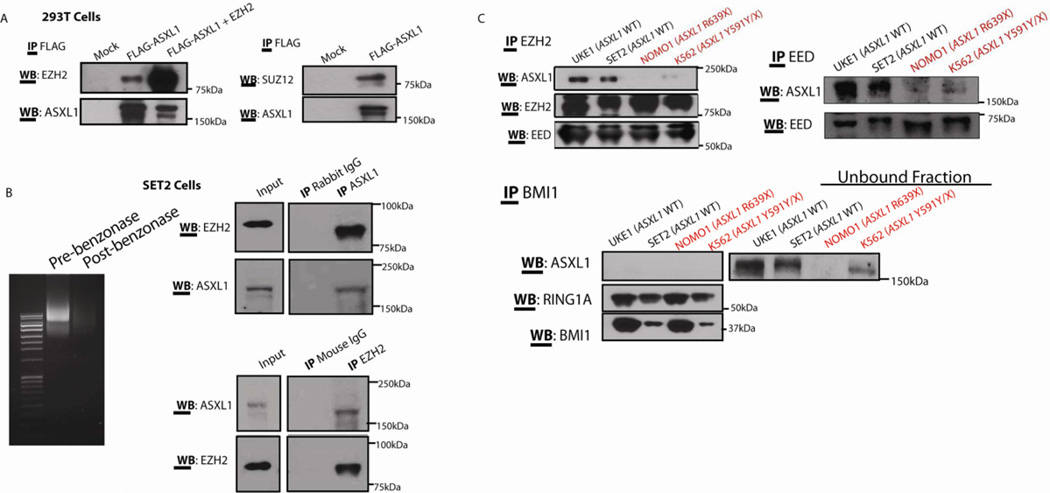
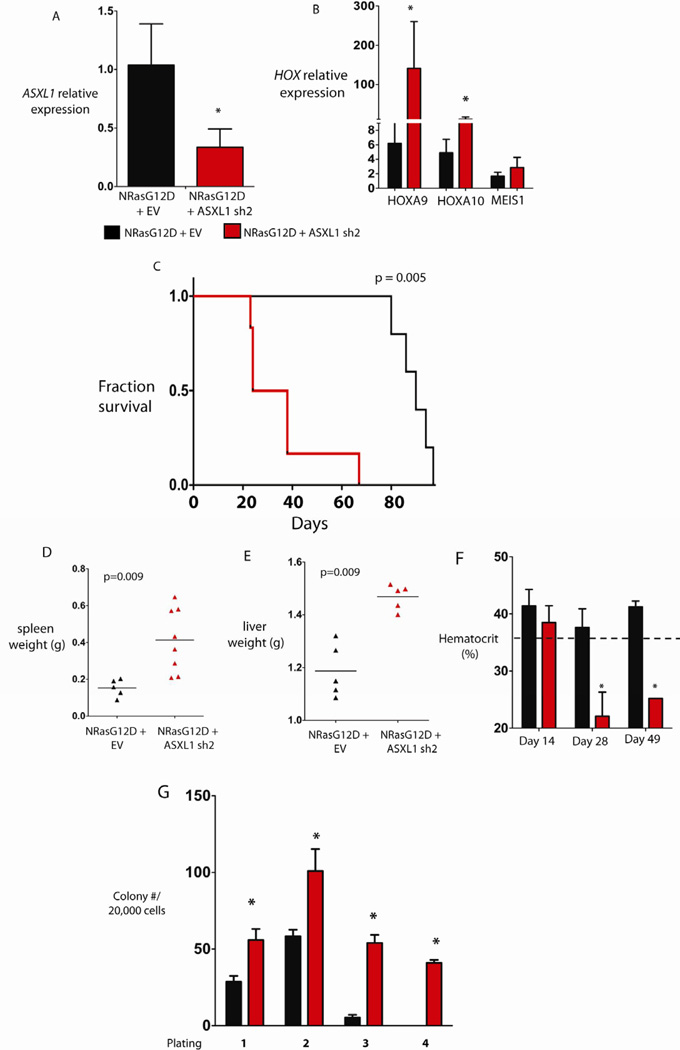
Recurrent somatic ASXL1 mutations occur in patients with myelodysplastic syndrome, myeloproliferative neoplasms, and acute myeloid leukemia, and are associated with adverse outcome. Despite the genetic and clinical data implicating ASXL1 mutations in myeloid malignancies, the mechanisms of transformation by ASXL1 mutations are not understood. Here, we identify that ASXL1 mutations result in loss of polycomb repressive complex 2 (PRC2)-mediated histone H3 lysine 27 (H3K27) tri-methylation. Through integration of microarray data with genome-wide histone modification ChIP-Seq data, we identify targets of ASXL1 repression, including the posterior HOXA cluster that is known to contribute to myeloid transformation. We demonstrate that ASXL1 associates with the PRC2, and that loss of ASXL1 in vivo collaborates with NRASG12D to promote myeloid leukemogenesis.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures
References
-
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
