

Sensitivity to TOP2 targeting chemotherapeutics is regulated by Oct1 and FILIP1L
- PMID: 22900064
- PMCID: PMC3416772
- DOI: 10.1371/journal.pone.0042921
Sensitivity to TOP2 targeting chemotherapeutics is regulated by Oct1 and FILIP1L
Abstract
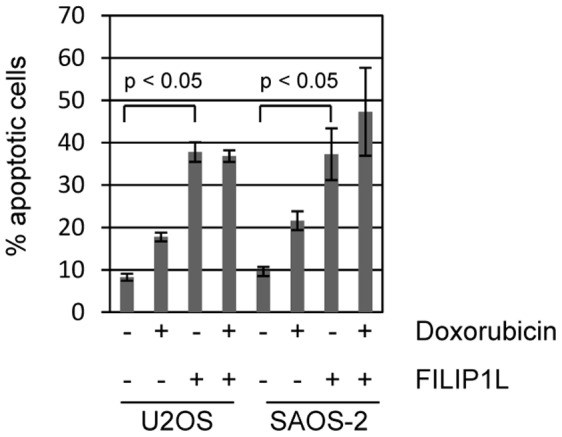
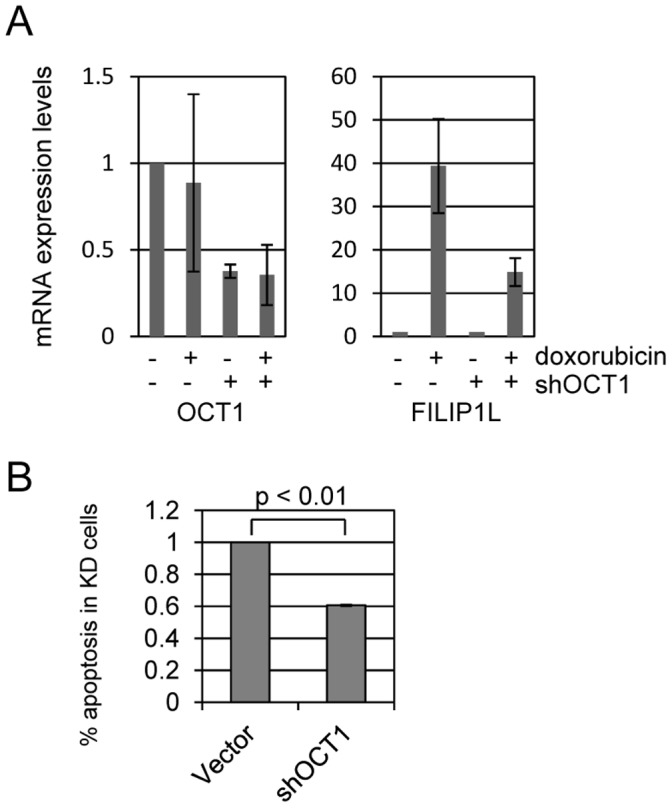
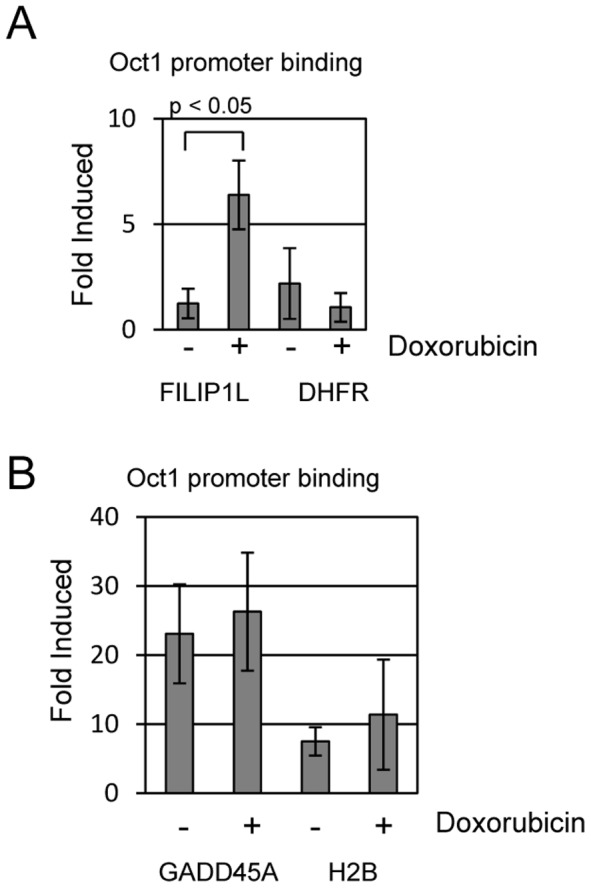
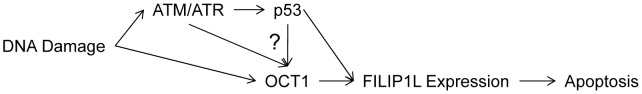
Topoisomerase II (TOP2) targeting drugs like doxorubicin and etoposide are frontline chemotherapeutics for a wide variety of solid and hematological malignancies, including breast and ovarian adenocarcinomas, lung cancers, soft tissue sarcomas, leukemias and lymphomas. These agents cause a block in DNA replication leading to a pronounced DNA damage response and initiation of apoptotic programs. Resistance to these agents is common, however, and elucidation of the mechanisms causing resistance to therapy could shed light on strategies to reduce the frequency of ineffective treatments. To explore these mechanisms, we utilized an unbiased shRNA screen to identify genes that regulate cell death in response to doxorubicin treatment. We identified the Filamin A interacting protein 1-like (FILIP1L) gene as a crucial mediator of apoptosis triggered by doxorubicin. FILIP1L shares significant similarity with bacterial SbcC, an ATPase involved in DNA repair. FILIP1L was originally described as DOC1, or "down-regulated in ovarian cancer" and has since been shown to be downregulated in a wide variety of human tumors. FILIP1L levels increase markedly through transcriptional mechanisms following treatment with doxorubicin and other TOP2 poisons, including etoposide and mitoxantrone, but not by the TOP2 catalytic inhibitors merbarone or dexrazoxane (ICRF187), or by UV irradiation. This induction requires the action of the OCT1 transcription factor, which relocalizes to the FILIP1L promoter and facilitates its expression following doxorubicin treatment. Our findings suggest that the FILIP1L expression status in tumors may influence the response to anti-TOP2 chemotherapeutics.
Conflict of interest statement
Figures
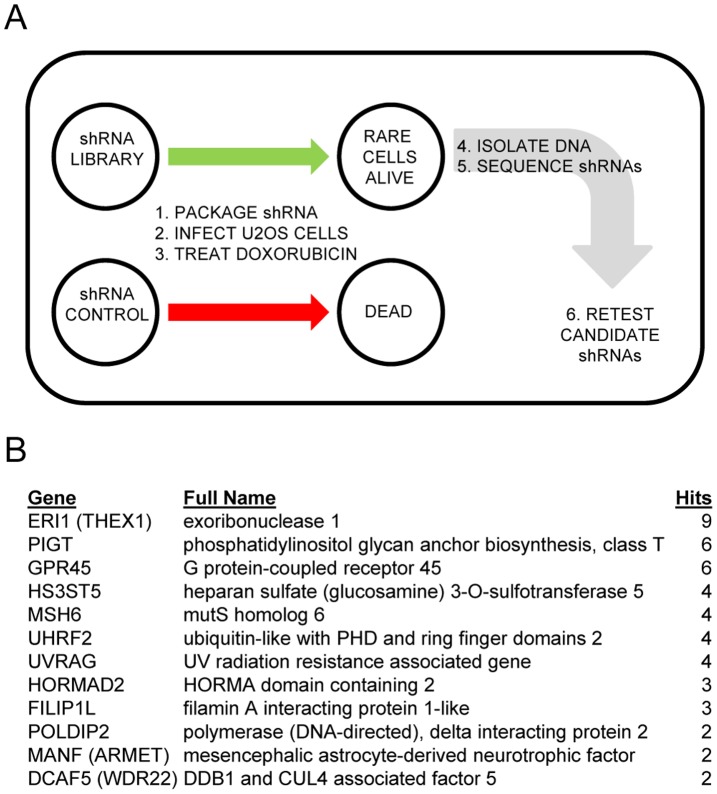
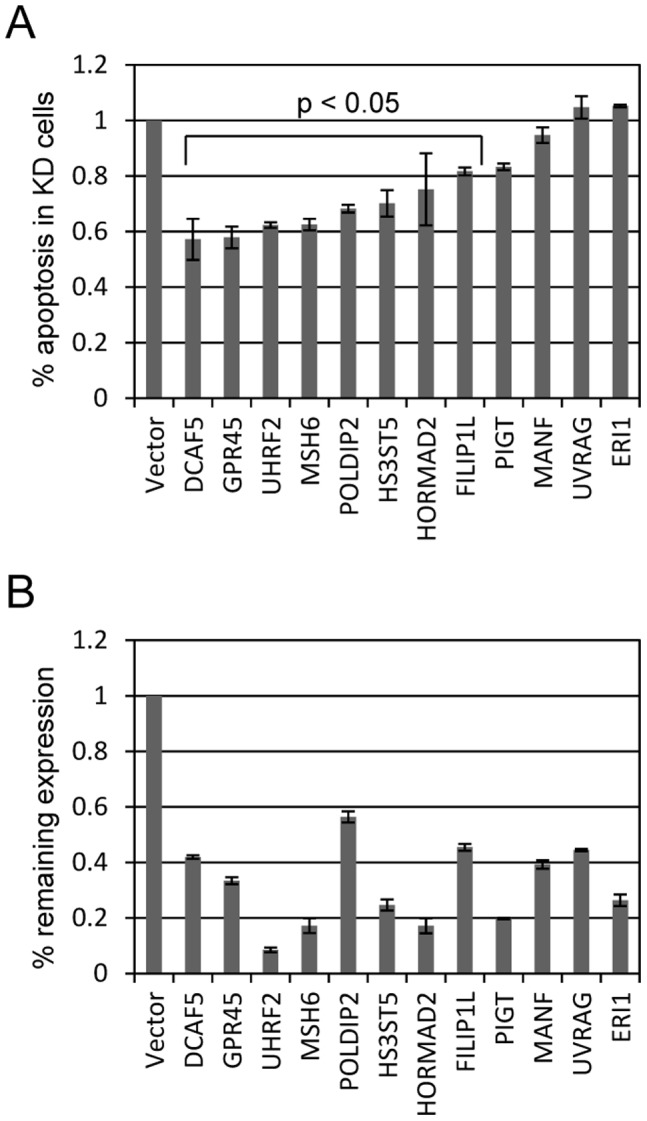
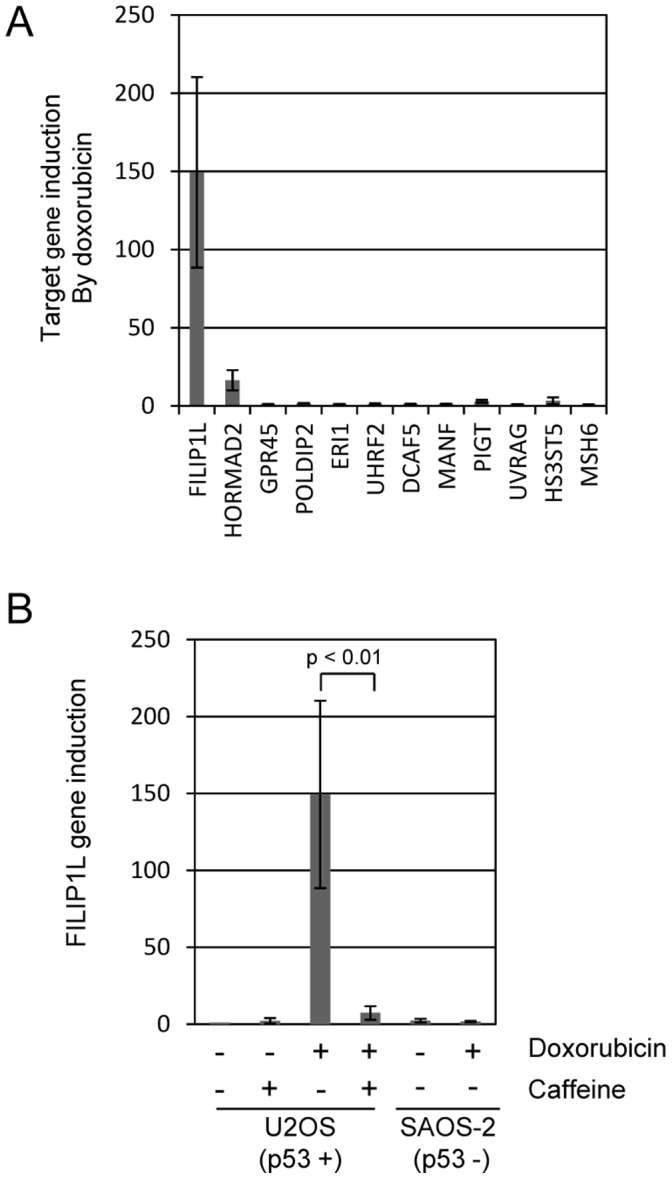
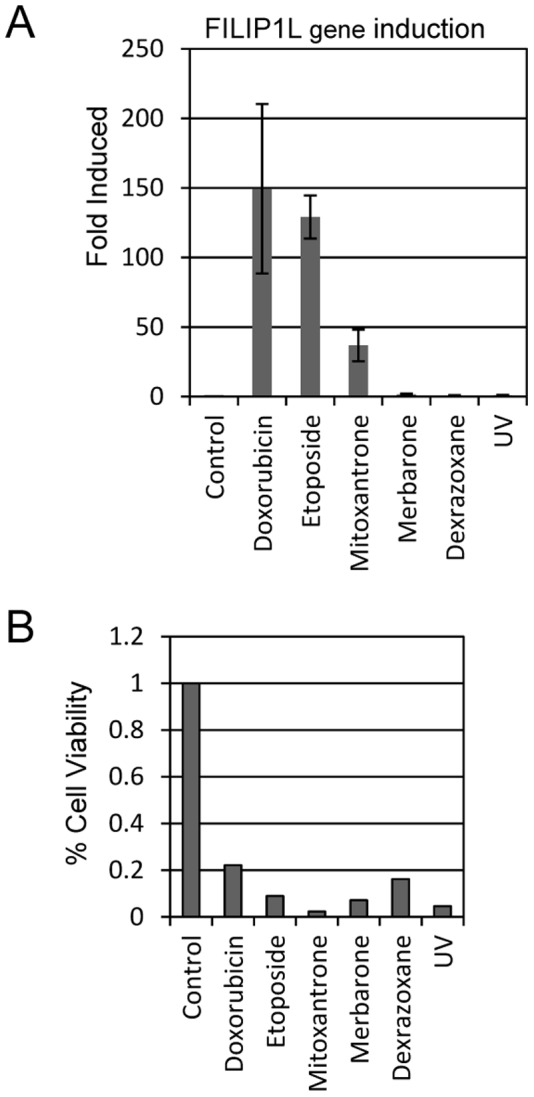
References
-
- Gros P, Ben Neriah Y, Croop JM, Housman DE (1986) Isolation and expression of a complementary DNA that confers multidrug resistance. Nature 323: 728–731. - PubMed
-
- Cole SP, Bhardwaj G, Gerlach JH, Mackie JE, Grant CE, et al. (1992) Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science 258: 1650–1654. - PubMed
-
- Gottesman MM, Pastan I (1993) Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu Rev Biochem 62: 385–427. - PubMed
-
- Kobayashi S, Boggon TJ, Dayaram T, Jänne PA, Kocher O, et al. (2005) EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med 352: 786–792. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
