

Transcriptome-based exon capture enables highly cost-effective comparative genomic data collection at moderate evolutionary scales
- PMID: 22900609
- PMCID: PMC3472323
- DOI: 10.1186/1471-2164-13-403
Transcriptome-based exon capture enables highly cost-effective comparative genomic data collection at moderate evolutionary scales
Abstract
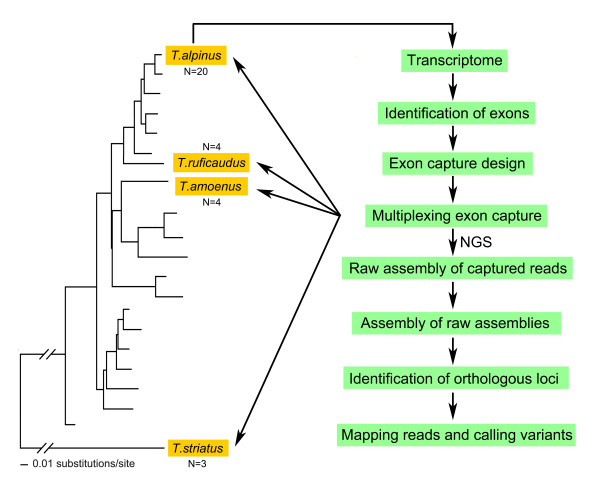
Background: To date, exon capture has largely been restricted to species with fully sequenced genomes, which has precluded its application to lineages that lack high quality genomic resources. We developed a novel strategy for designing array-based exon capture in chipmunks (Tamias) based on de novo transcriptome assemblies. We evaluated the performance of our approach across specimens from four chipmunk species.
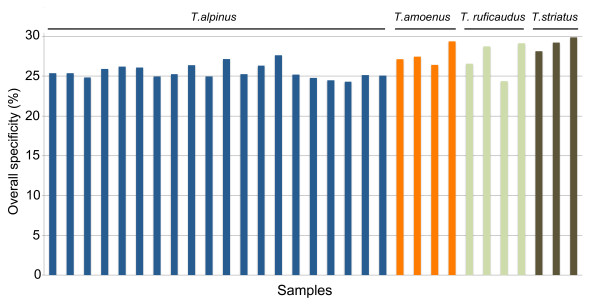
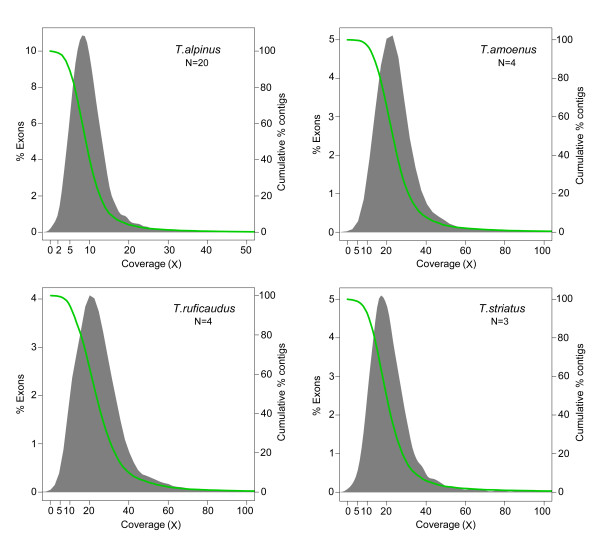
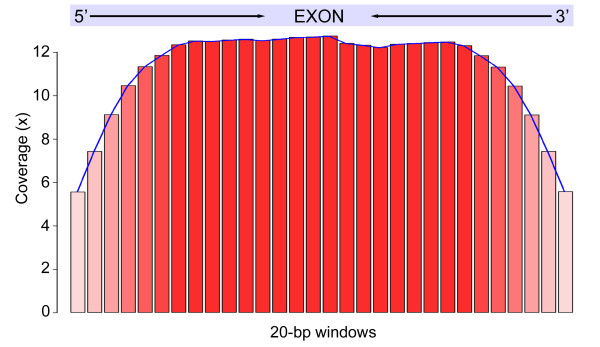
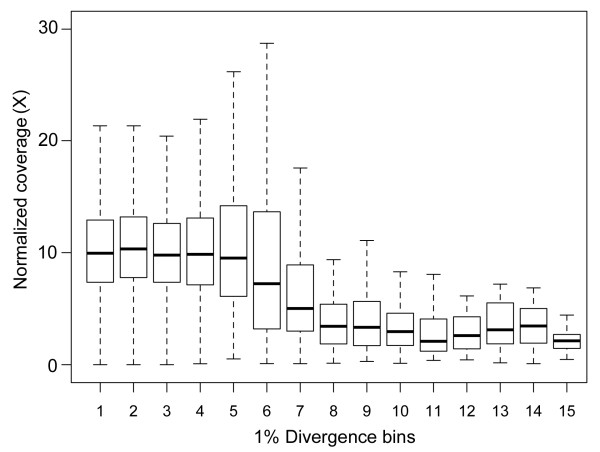
Results: We selectively targeted 11,975 exons (~4 Mb) on custom capture arrays, and enriched over 99% of the targets in all libraries. The percentage of aligned reads was highly consistent (24.4-29.1%) across all specimens, including in multiplexing up to 20 barcoded individuals on a single array. Base coverage among specimens and within targets in each species library was uniform, and the performance of targets among independent exon captures was highly reproducible. There was no decrease in coverage among chipmunk species, which showed up to 1.5% sequence divergence in coding regions. We did observe a decline in capture performance of a subset of targets designed from a much more divergent ground squirrel genome (30 My), however, over 90% of the targets were also recovered. Final assemblies yielded over ten thousand orthologous loci (~3.6 Mb) with thousands of fixed and polymorphic SNPs among species identified.
Conclusions: Our study demonstrates the potential of a transcriptome-enabled, multiplexed, exon capture method to create thousands of informative markers for population genomic and phylogenetic studies in non-model species across the tree of life.
Figures
References
-
- Good JM. Reduced representation methods for subgenomic enrichment and next-generation sequencing. Methods Mol Biol. 2011;772:85–103. - PubMed
-
- Yi X, Liang Y, Huerta-Sanchez E, Jin X, Cuo ZX, Pool JE, Xu X, Jiang H, Vinckenbosch N, Korneliussen TS, Zheng H, Liu T, He W, Li K, Luo R, Nie X, Wu H, Zhao M, Cao H, Zou J, Shan Y, Li S, Yang Q, Asan, Ni P, Tian G, Xu J, Liu X, Jiang T, Wu R, Zhou G, Tang M, Qin J, Wang T, Feng S, Li G, Huasang, Luosang J, Wang W, Chen F, Wang Y, Zheng X, Li Z, Bianba Z, Yang G, Wang X, Tang S, Gao G, Chen Y, Luo Z, Gusang L, Cao Z, Zhang Q, Ouyang W, Ren X, Liang H, Zheng H, Huang Y, Li J, Bolund L, Kristiansen K, Li Y, Zhang Y, Zhang X, Li R, Li S, Yang H, Nielsen R, Wang J, Wang J. Sequencing of 50 human exomes reveals adaptation to high altitude. Science. 2010;329:75–78. doi: 10.1126/science.1190371. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
