

Reawakening fetal hemoglobin: prospects for new therapies for the β-globin disorders
- PMID: 22904296
- PMCID: PMC4467860
- DOI: 10.1182/blood-2012-06-292078
Reawakening fetal hemoglobin: prospects for new therapies for the β-globin disorders
Abstract
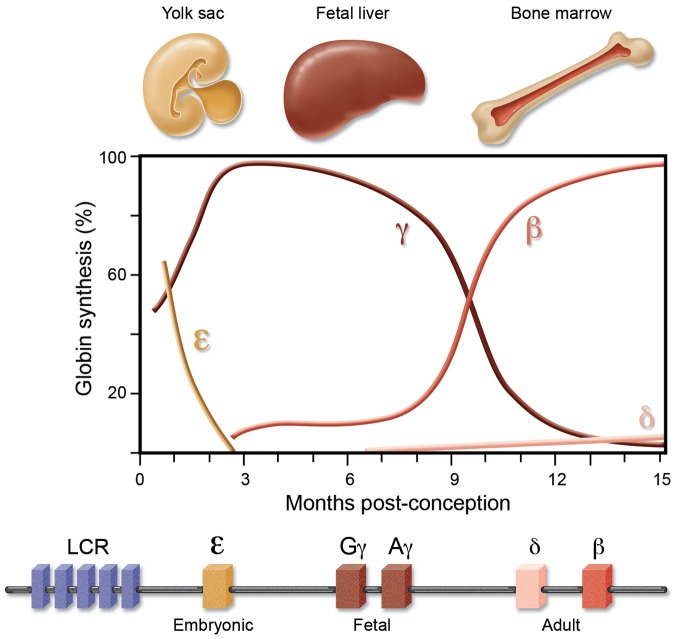
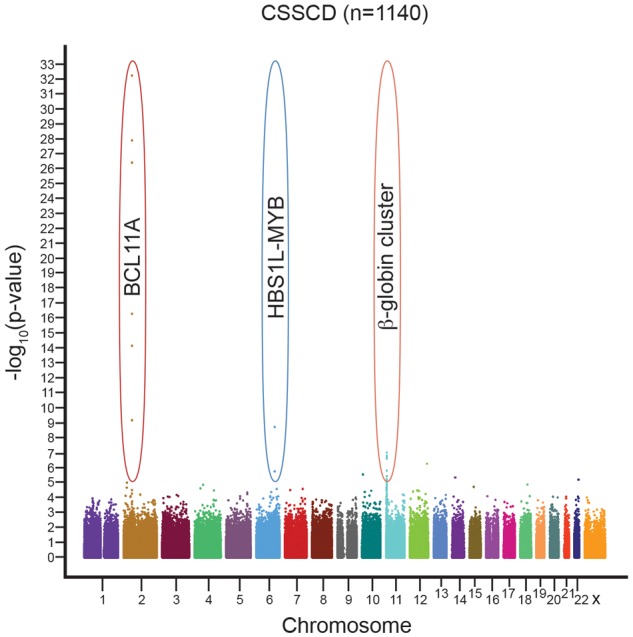
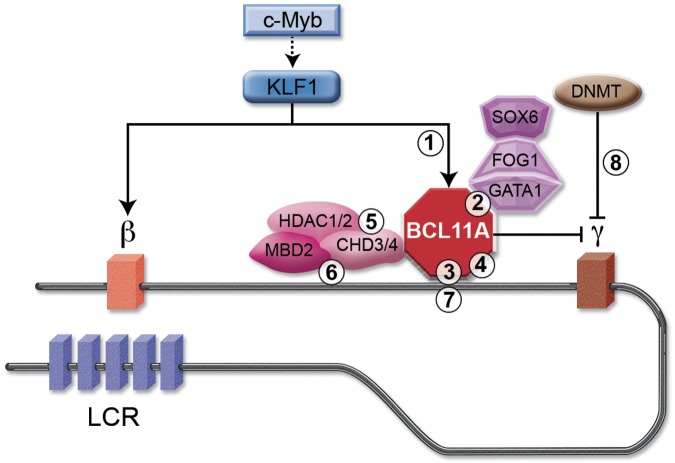
The level of fetal hemoglobin (HbF) modifies the severity of the common β-globin disorders. Knowledge of the normal mechanisms that repress HbF in the adult stage has remained limited until recently despite nearly 3 decades of molecular investigation, in part because of imperfect model systems. Recent studies have provided new insights into the developmental regulation of globin genes and identified specific transcription factors and epigenetic regulators responsible for physiologic silencing of HbF. Most prominent among these regulators is BCL11A, a transcriptional repressor that inhibits adult-stage HbF expression. KLF1 and c-Myb are additional critical HbF-regulating erythroid transcription factors more broadly involved in erythroid gene expression programs. Chromatin modifiers, including histone deacetylases and DNA methyltransferases, also play key roles in orchestrating appropriate globin gene expression. Taken together, these discoveries present novel therapeutic targets for further consideration. Although substantial hurdles remain, opportunities are now rich for the rational design of HbF inducers.
Figures
References
-
- Kauf TL, Coates TD, Huazhi L, Mody-Patel N, Hartzema AG. The cost of health care for children and adults with sickle cell disease. Am J Hematol. 2009;84(6):323–327. - PubMed
-
- World urbanization prospects, the 2011 revision. 2012. [Accessed May 15, 2012]. http://esa.un.org/unup/documentation/highlights.htm.
-
- Pauling L, Itano HA. Sickle cell anemia a molecular disease. Science. 1949;110(2865):543–548. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
