

Predicting HLA class I non-permissive amino acid residues substitutions
- PMID: 22905104
- PMCID: PMC3414483
- DOI: 10.1371/journal.pone.0041710
Predicting HLA class I non-permissive amino acid residues substitutions
Abstract
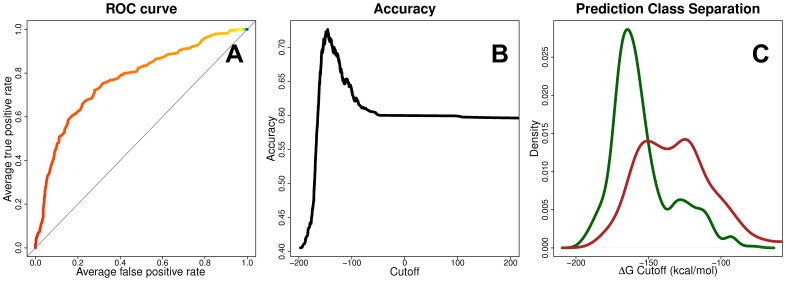
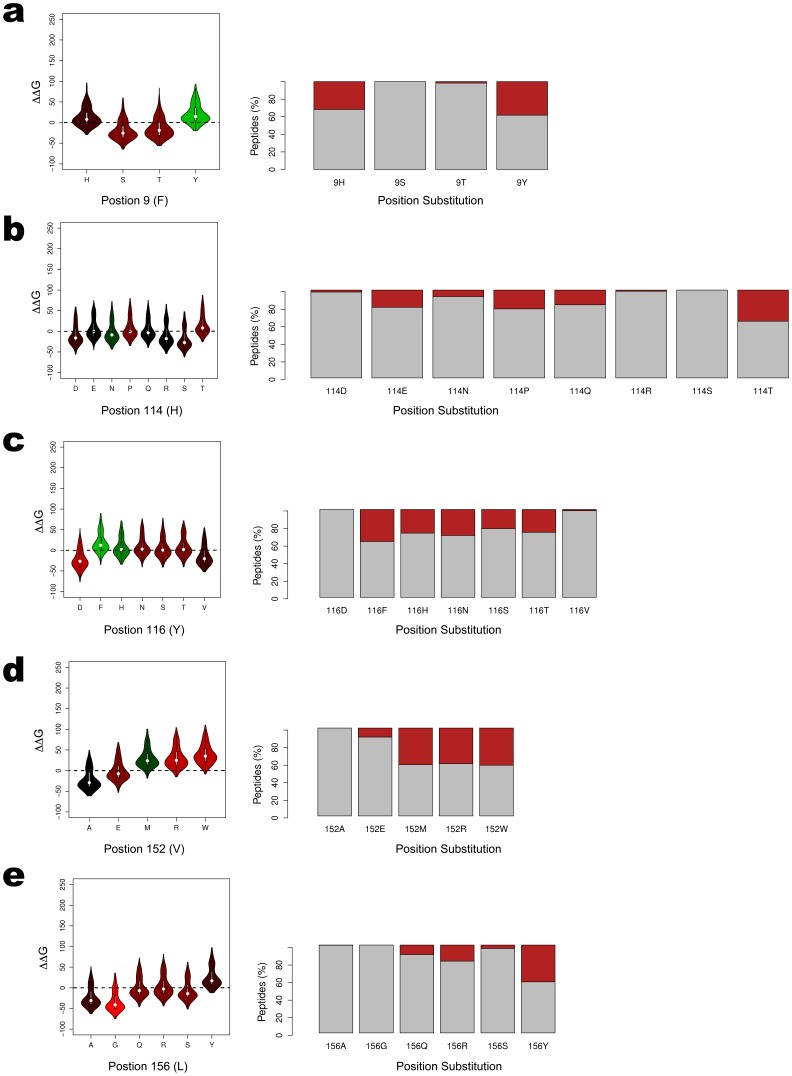
Prediction of peptide binding to human leukocyte antigen (HLA) molecules is essential to a wide range of clinical entities from vaccine design to stem cell transplant compatibility. Here we present a new structure-based methodology that applies robust computational tools to model peptide-HLA (p-HLA) binding interactions. The method leverages the structural conservation observed in p-HLA complexes to significantly reduce the search space and calculate the system's binding free energy. This approach is benchmarked against existing p-HLA complexes and the prediction performance is measured against a library of experimentally validated peptides. The effect on binding activity across a large set of high-affinity peptides is used to investigate amino acid mismatches reported as high-risk factors in hematopoietic stem cell transplantation.
Conflict of interest statement
Figures
References
-
- Little AM, Parham P (1999) Polymorphism and evolution of HLA class I and II genes and molecules. Rev Immunogenet 1: 105–123. - PubMed
-
- Shlomchik WD (2007) Graft-versus-host disease. Nat Rev Immunol 7: 340–352. - PubMed
-
- Gao GF, Tormo J, Gerth UC, Wyer JR, McMichael AJ, et al. (1997) Crystal structure of the complex between human CD8alpha(alpha) and HLA-A2. Nature 387: 630–634. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
