

Sustained NMDA receptor activation by spreading depolarizations can initiate excitotoxic injury in metabolically compromised neurons
- PMID: 22907056
- PMCID: PMC3528997
- DOI: 10.1113/jphysiol.2012.234476
Sustained NMDA receptor activation by spreading depolarizations can initiate excitotoxic injury in metabolically compromised neurons
Abstract
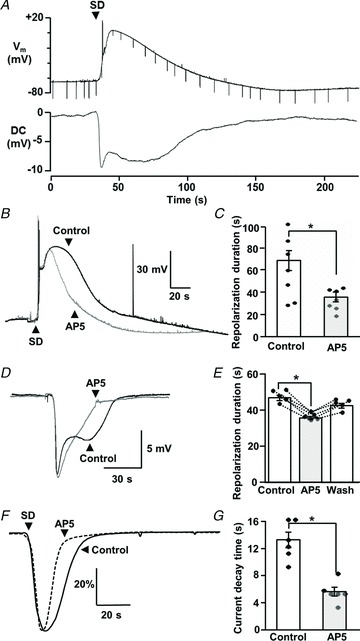
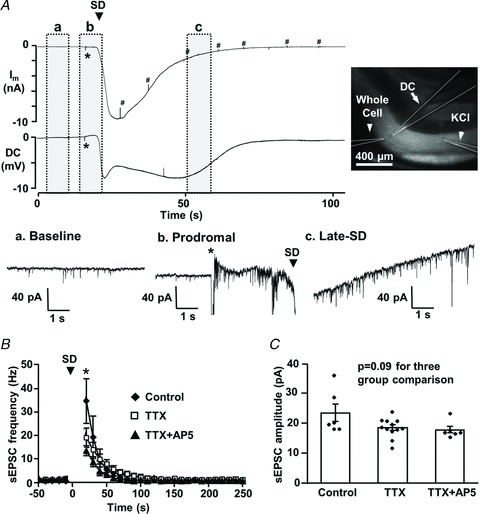
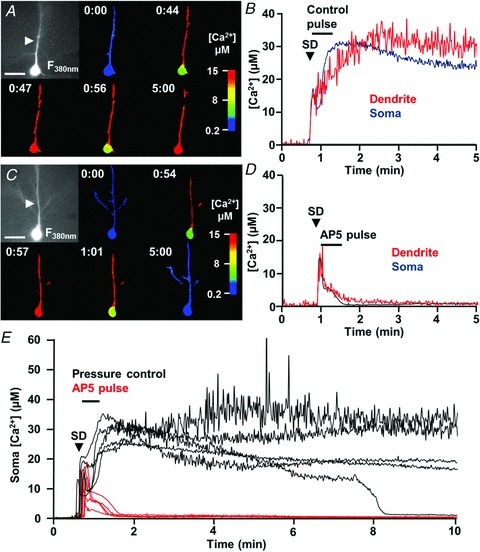
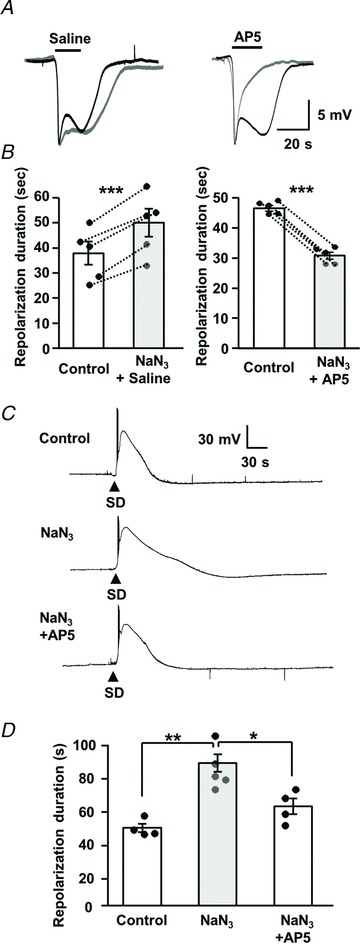
Spreading depolarizations (SDs) are slowly propagating waves of near-complete neuronal and glial depolarization. SDs have been recorded in patients with brain injury, and the incidence of SD significantly correlates with outcome severity. Although it is well accepted that the ionic dyshomeostasis of SD presents a severe metabolic burden, there is currently limited understanding of SD-induced injury processes at a cellular level. In the current study we characterized events accompanying SD in the hippocampal CA1 region of murine brain slices, using whole-cell recordings and single-cell Ca(2+) imaging. We identified an excitatory phase that persisted for approximately 2 min following SD onset, and accompanied with delayed dendritic ionic dyshomeostasis. The excitatory phase coincided with a significant increase in presynaptic glutamate release, evidenced by a transient increase in spontaneous EPSC frequency and paired-pulse depression of evoked EPSCs. Activation of NMDA receptors (NMDARs) during this late excitatory phase contributed to the duration of individual neuronal depolarizations and delayed recovery of extracellular slow potential changes. Selectively targeting the NMDAR activation following SD onset (by delayed pressure application of a competitive NMDAR antagonist) significantly decreased the duration of cellular depolarizations. Recovery of dendritic Ca(2+) elevations following SD were also sensitive to delayed NMDA antagonist application. Partial inhibition of neuronal energy metabolism converted SD into an irrecoverable event with persistent Ca(2+) overload and membrane compromise. Delayed NMDAR block was sufficient to prevent these acute injurious events in metabolically compromised neurons. These results identify a significant contribution of a late component of SD that could underlie neuronal injury in pathological circumstances.
Figures




References
-
- Anderson TR, Andrew RD. Spreading depression: imaging and blockade in the rat neocortical brain slice. J Neurophysiol. 2002;88:2713–2725. - PubMed
-
- Blatow M, Caputi A, Burnashev N, Monyer H, Rozov A. Ca2+ buffer saturation underlies paired pulse facilitation in calbindin-D28k-containing terminals. Neuron. 2003;38:79–88. - PubMed
-
- Canals S, Makarova I, Lopez-Aguado L, Largo C, Ibarz JM, Herreras O. Longitudinal depolarization gradients along the somatodendritic axis of CA1 pyramidal cells: a novel feature of spreading depression. J Neurophysiol. 2005;94:943–951. - PubMed
-
- Connor JA, Cormier RJ. Cumulative effects of glutamate microstimulation on Ca2+ responses of CA1 hippocampal pyramidal neurons in slice. J Neurophysiol. 2000;83:90–98. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous