

Molecular basis for negative regulation of the glucagon receptor
- PMID: 22908259
- PMCID: PMC3437825
- DOI: 10.1073/pnas.1206734109
Molecular basis for negative regulation of the glucagon receptor
Abstract
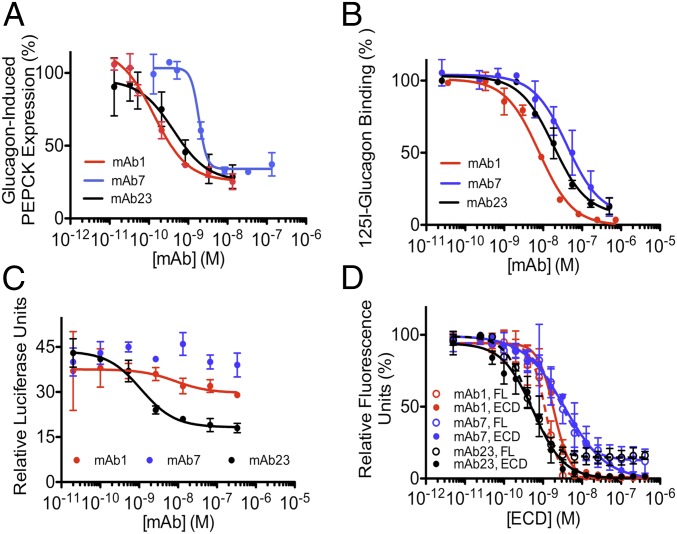
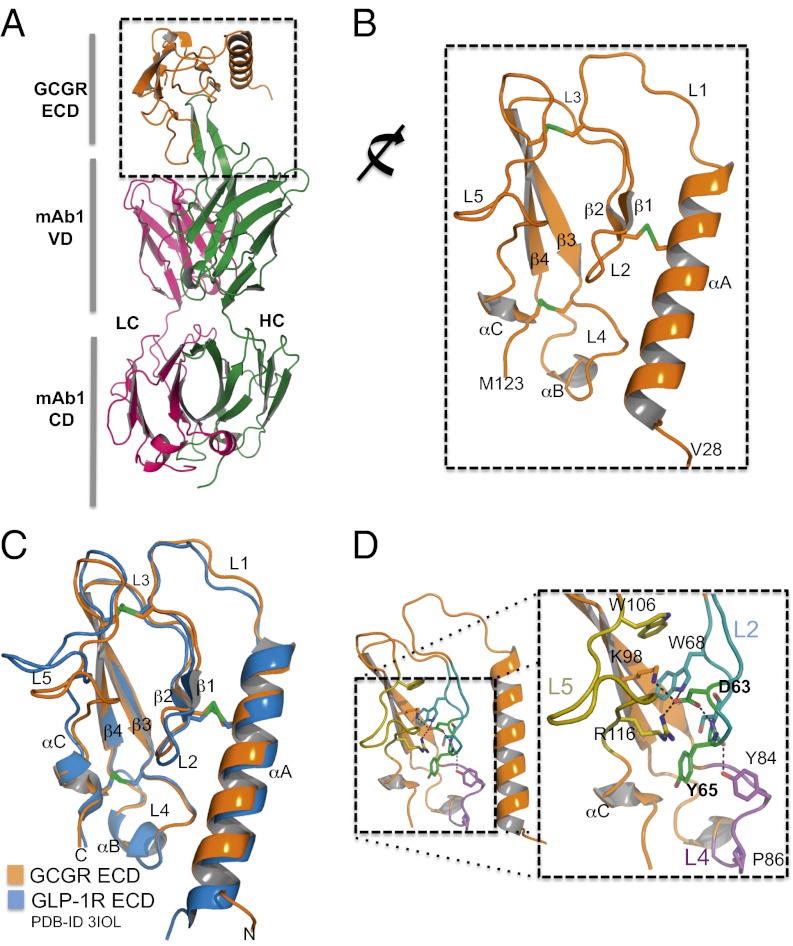
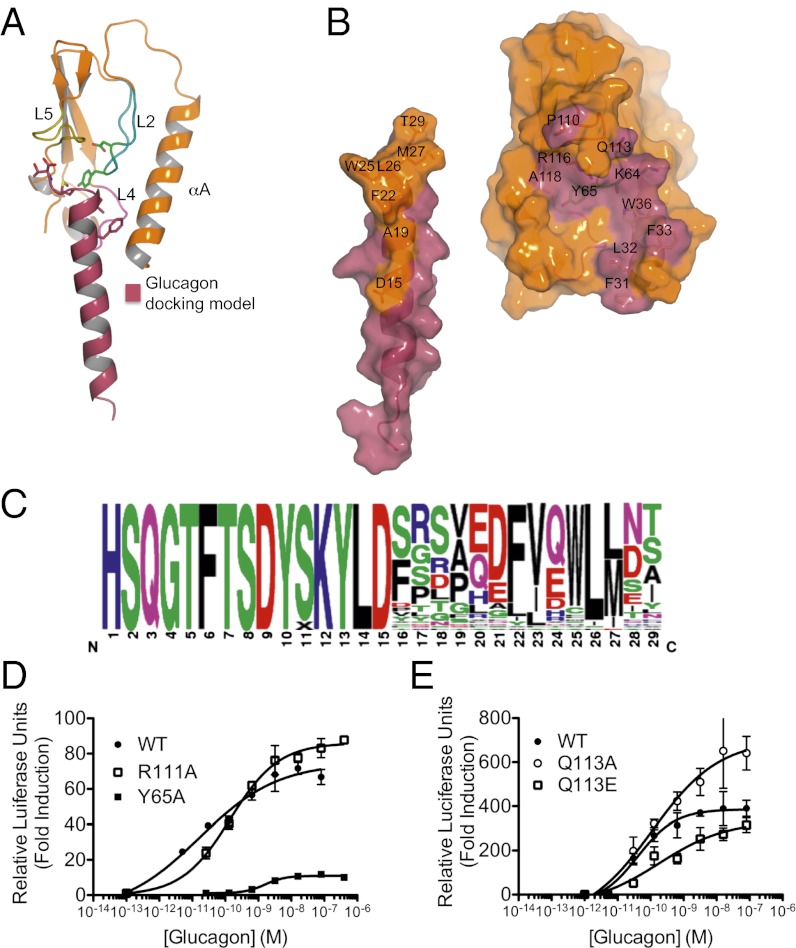
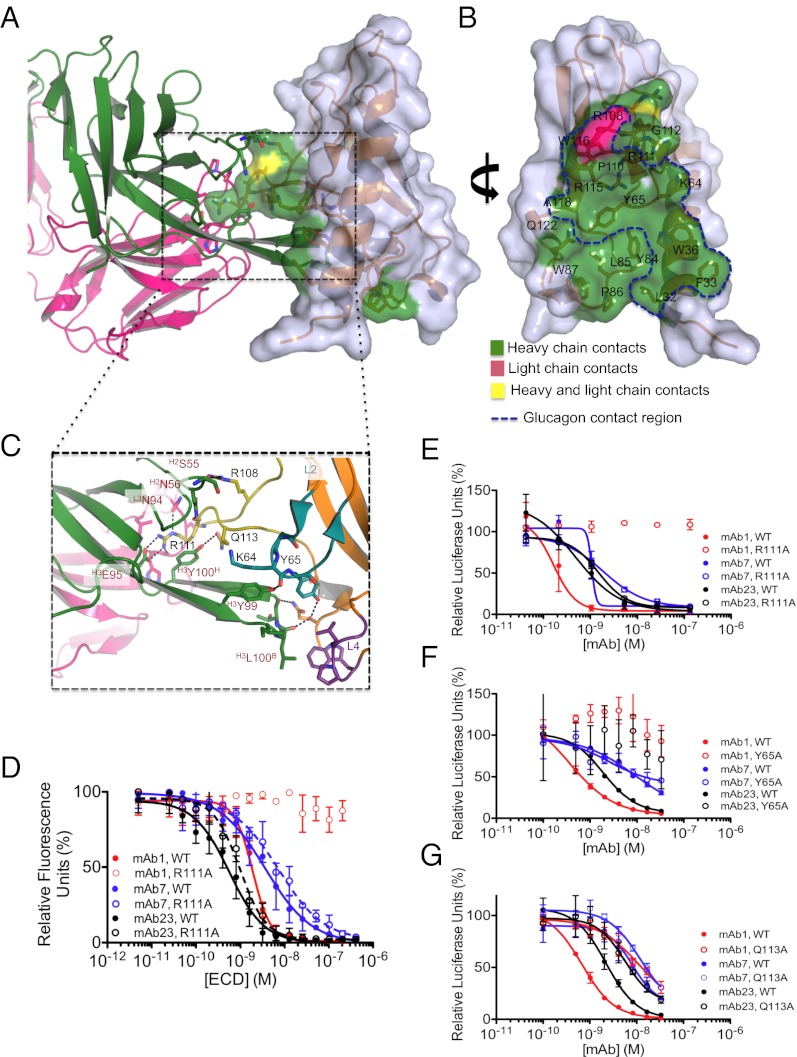
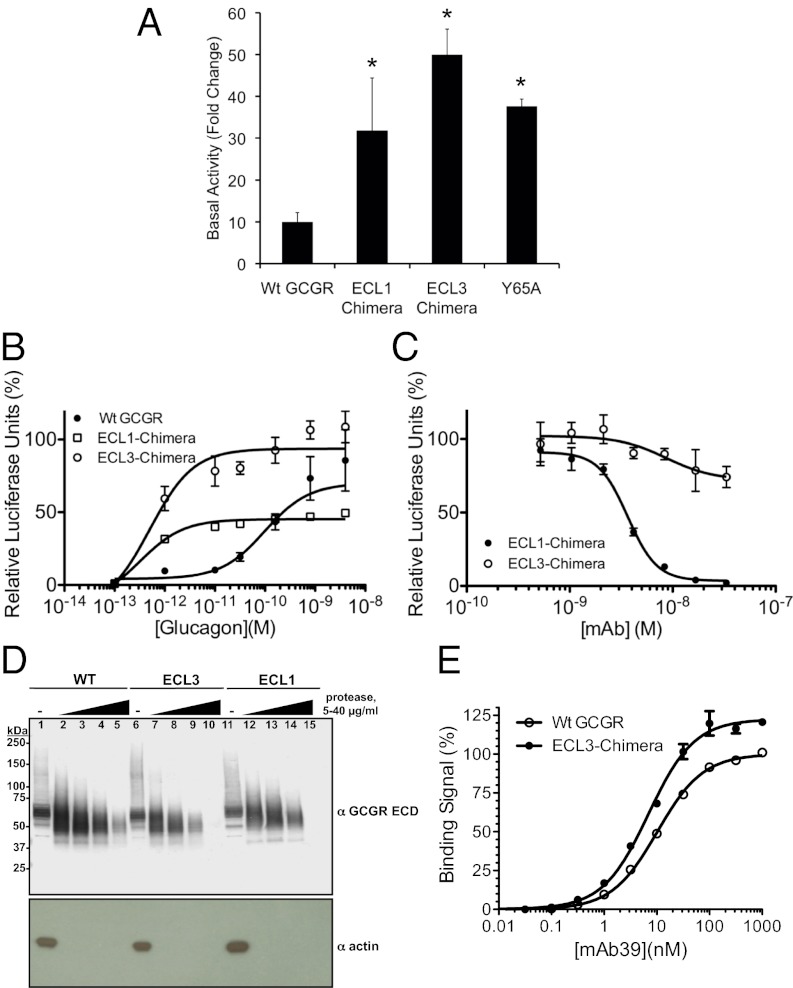
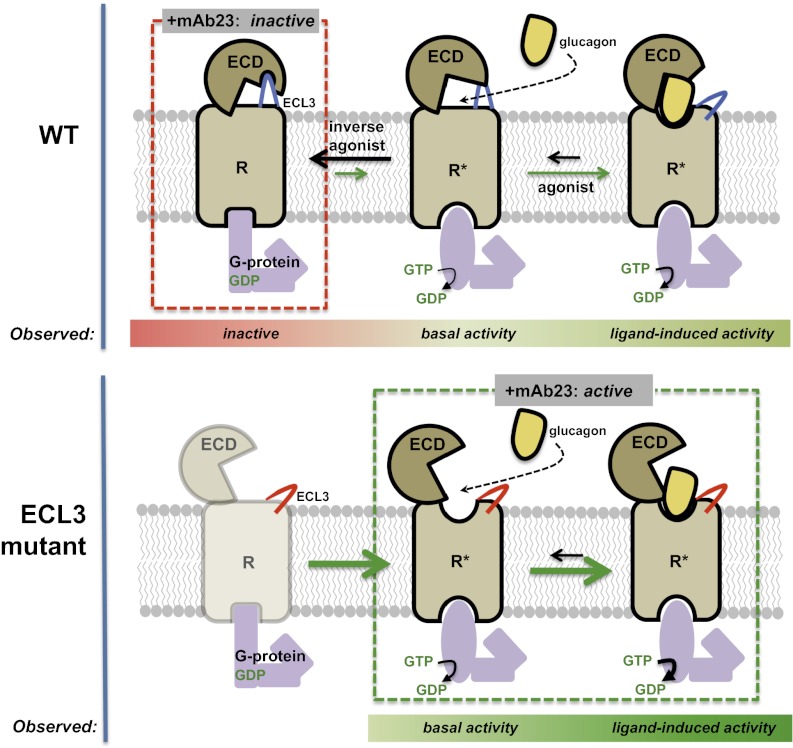
Members of the class B family of G protein-coupled receptors (GPCRs) bind peptide hormones and have causal roles in many diseases, ranging from diabetes and osteoporosis to anxiety. Although peptide, small-molecule, and antibody inhibitors of these GPCRs have been identified, structure-based descriptions of receptor antagonism are scarce. Here we report the mechanisms of glucagon receptor inhibition by blocking antibodies targeting the receptor's extracellular domain (ECD). These studies uncovered a role for the ECD as an intrinsic negative regulator of receptor activity. The crystal structure of the ECD in complex with the Fab fragment of one antibody, mAb1, reveals that this antibody inhibits glucagon receptor by occluding a surface extending across the entire hormone-binding cleft. A second antibody, mAb23, blocks glucagon binding and inhibits basal receptor activity, indicating that it is an inverse agonist and that the ECD can negatively regulate receptor activity independent of ligand binding. Biochemical analyses of receptor mutants in the context of a high-resolution ECD structure show that this previously unrecognized inhibitory activity of the ECD involves an interaction with the third extracellular loop of the receptor and suggest that glucagon-mediated structural changes in the ECD accompany receptor activation. These studies have implications for the design of drugs to treat class B GPCR-related diseases, including the potential for developing novel allosteric regulators that target the ECDs of these receptors.
Conflict of interest statement
Conflict of interest statement: All authors are employees of Genentech Inc.
Figures
References
-
- Jelinek LJ, et al. Expression cloning and signaling properties of the rat glucagon receptor. Science. 1993;259:1614–1616. - PubMed
-
- Dallas-Yang Q, et al. Hepatic glucagon receptor binding and glucose-lowering in vivo by peptidyl and non-peptidyl glucagon receptor antagonists. Eur J Pharmacol. 2004;501:225–234. - PubMed
-
- Petersen KF, Sullivan JT. Effects of a novel glucagon receptor antagonist (Bay 27-9955) on glucagon-stimulated glucose production in humans. Diabetologia. 2001;44:2018–2024. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
