

MyD88 inhibition amplifies dendritic cell capacity to promote pancreatic carcinogenesis via Th2 cells
- PMID: 22908323
- PMCID: PMC3428946
- DOI: 10.1084/jem.20111706
MyD88 inhibition amplifies dendritic cell capacity to promote pancreatic carcinogenesis via Th2 cells
Abstract
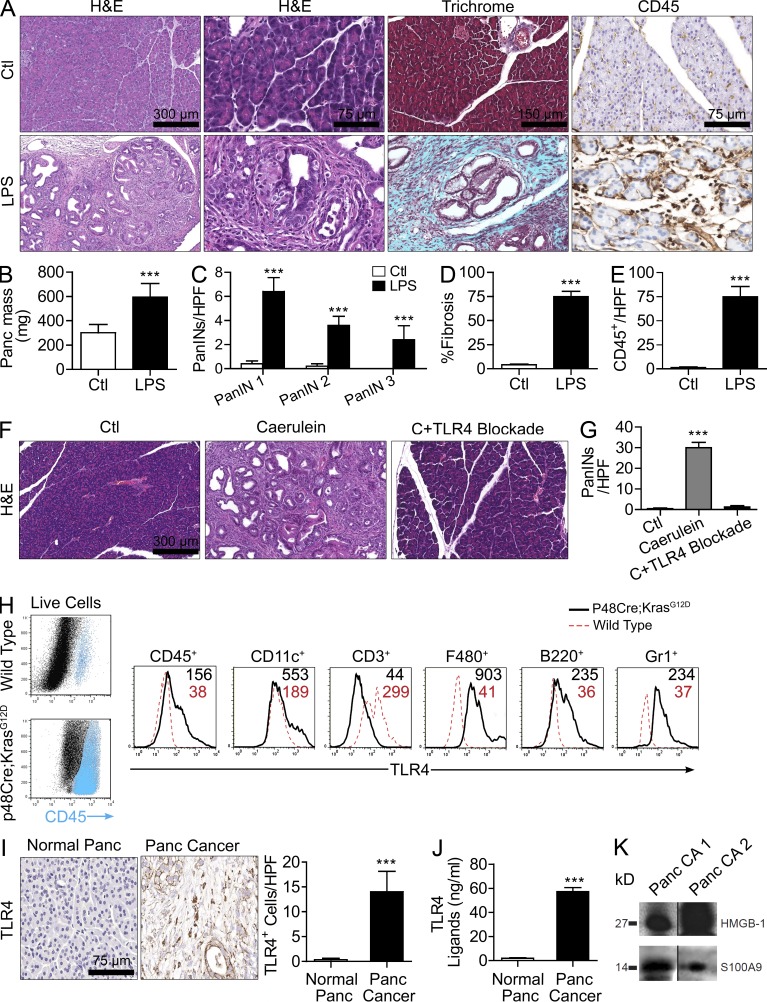
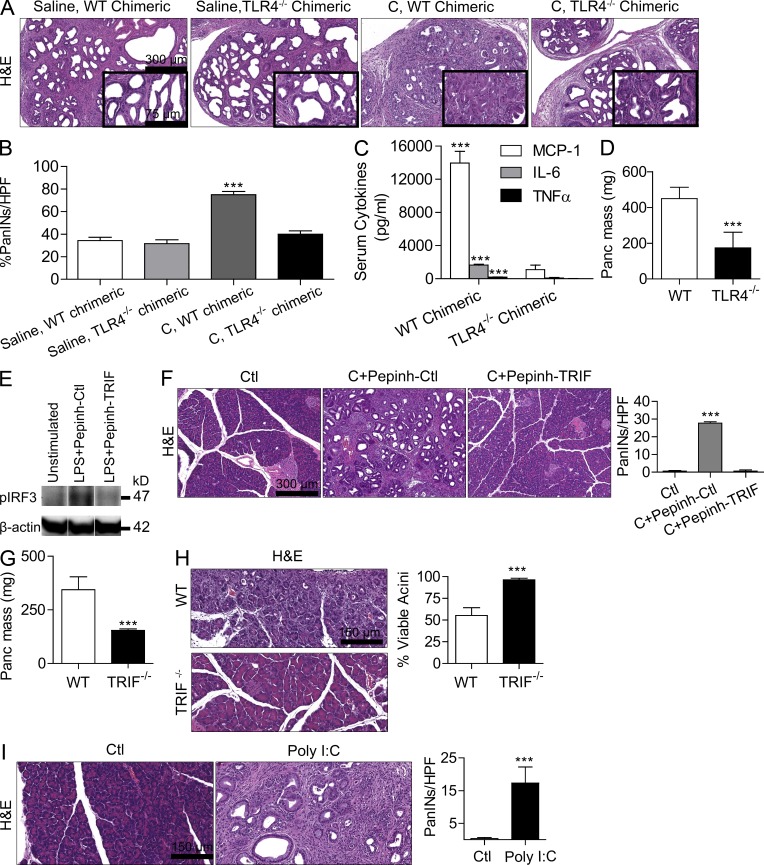
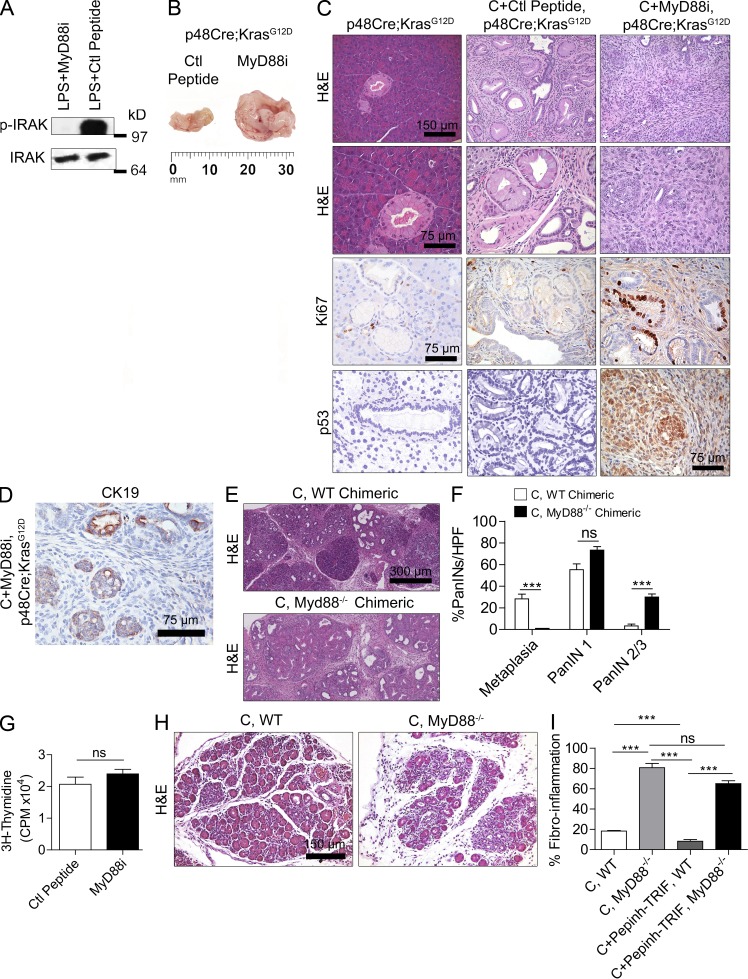
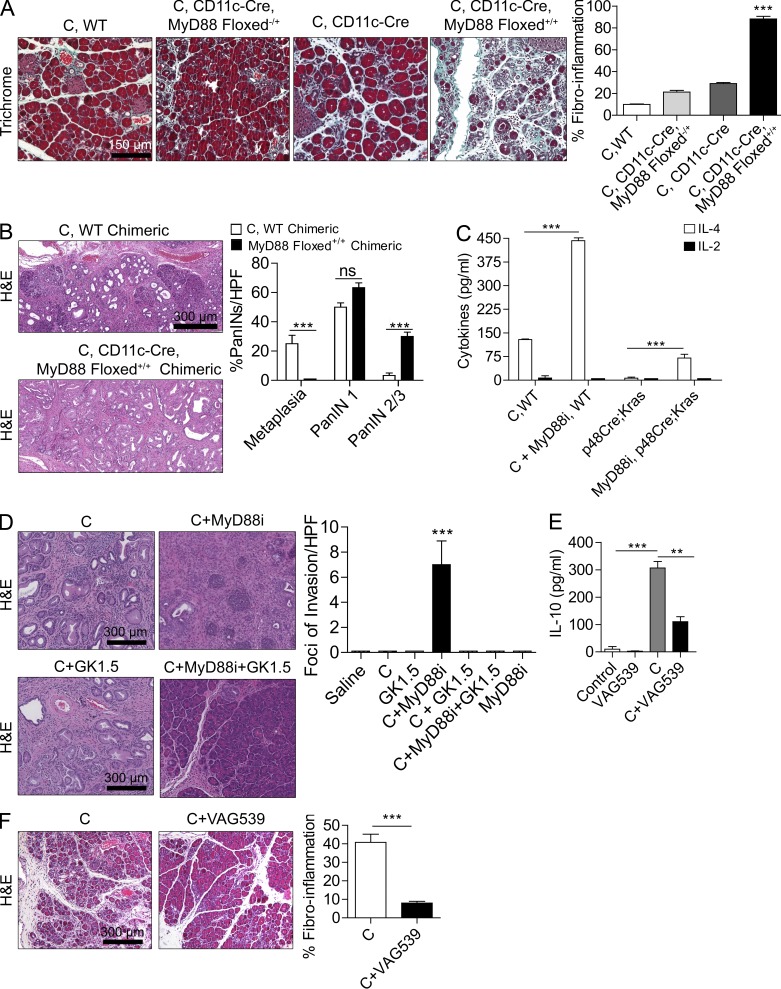
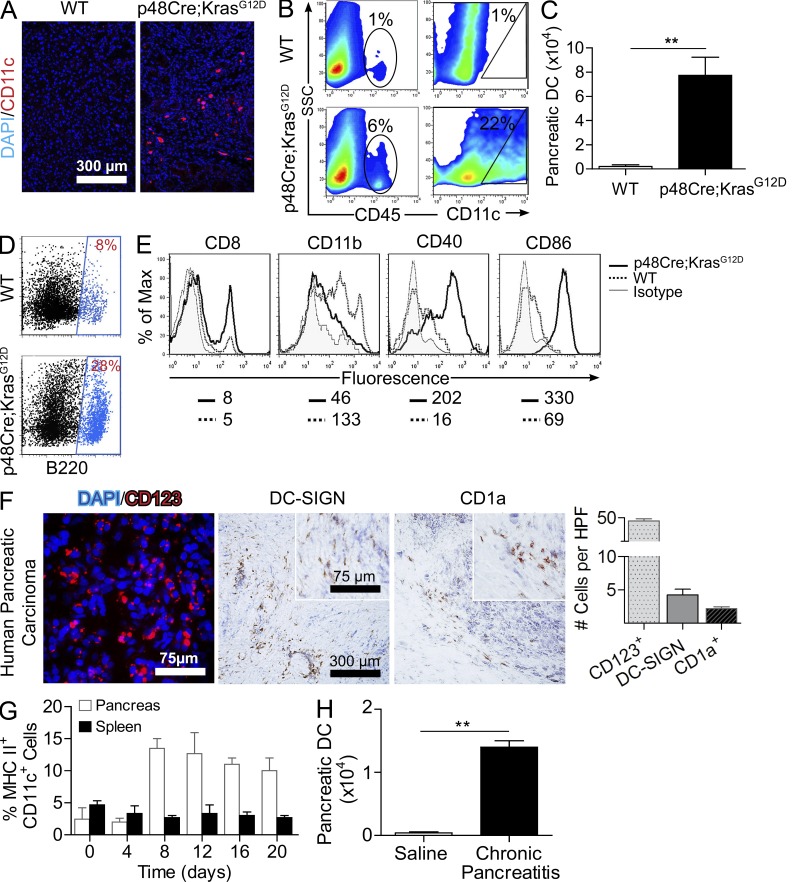
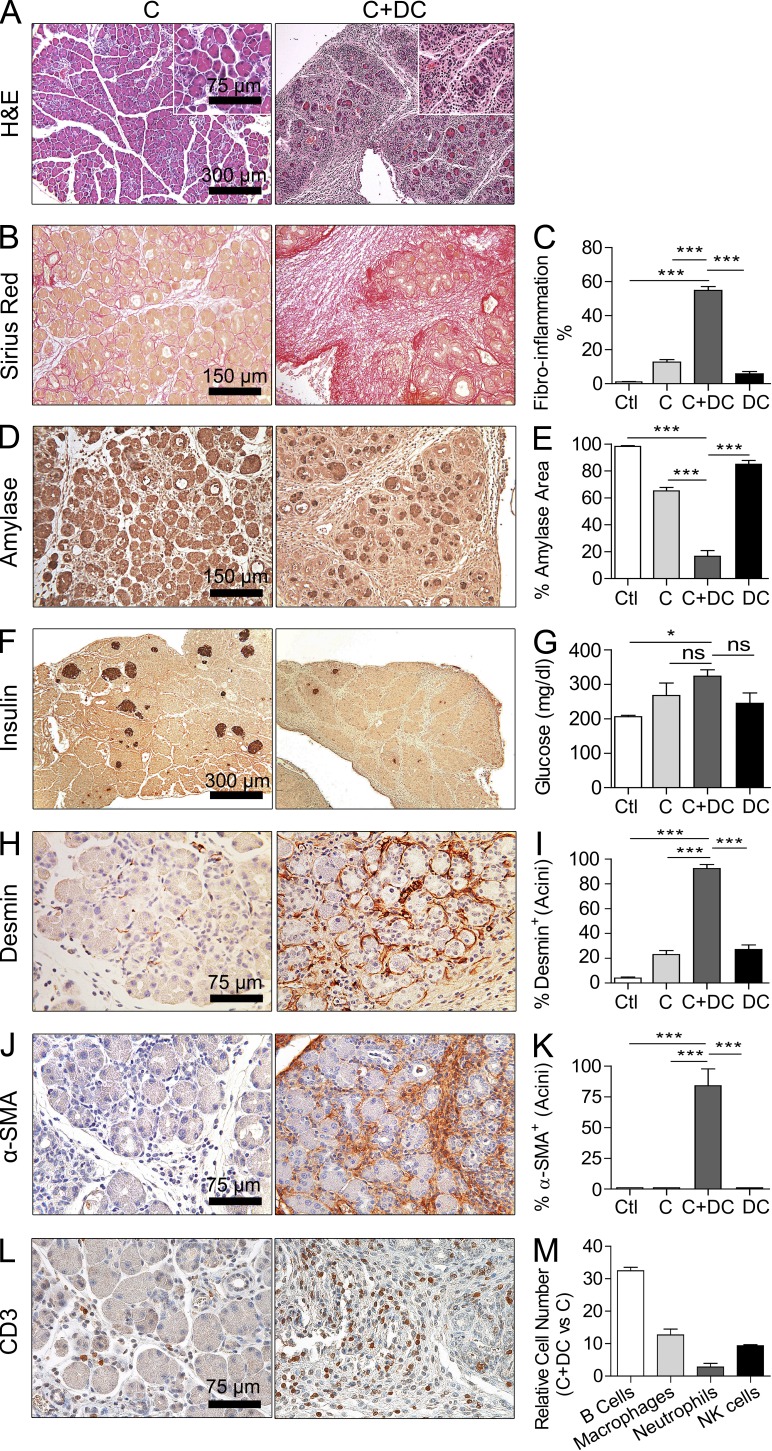
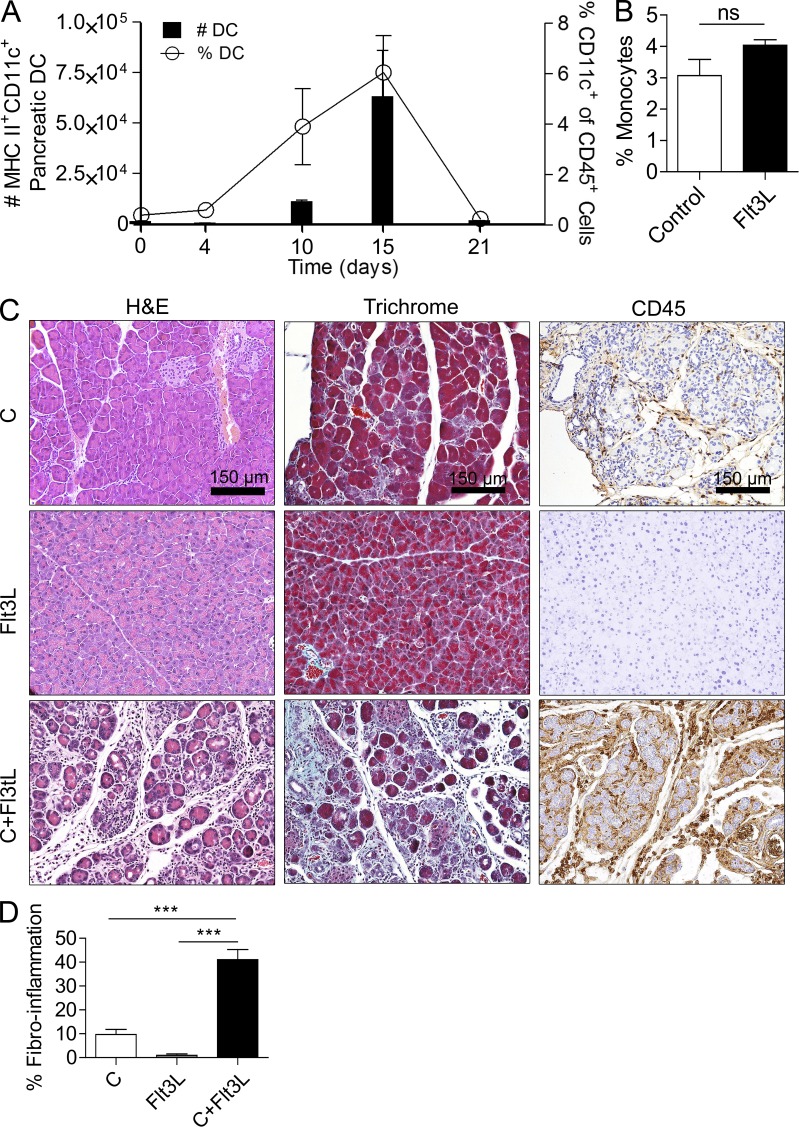
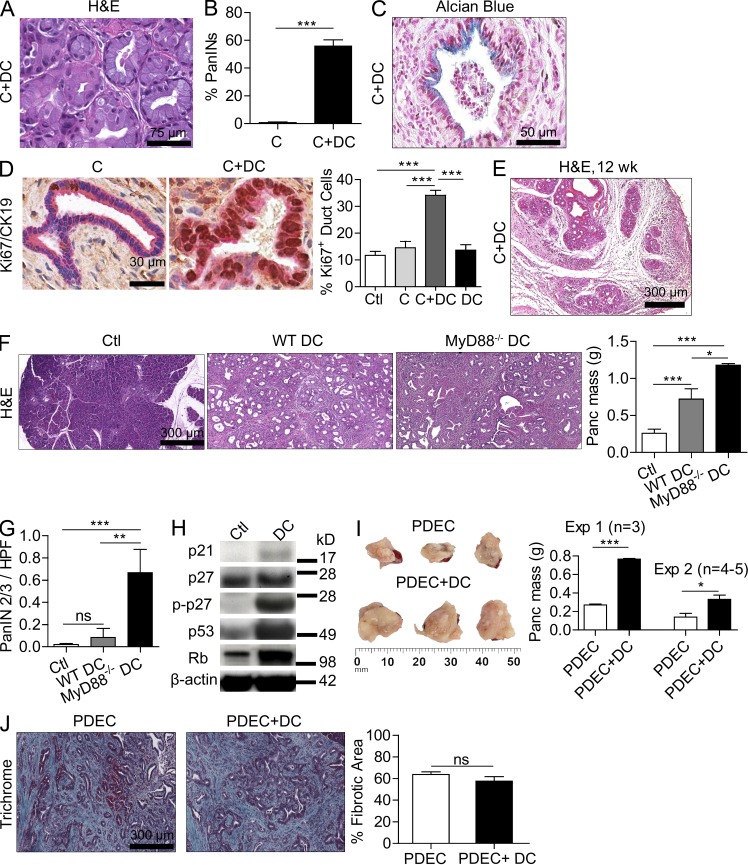
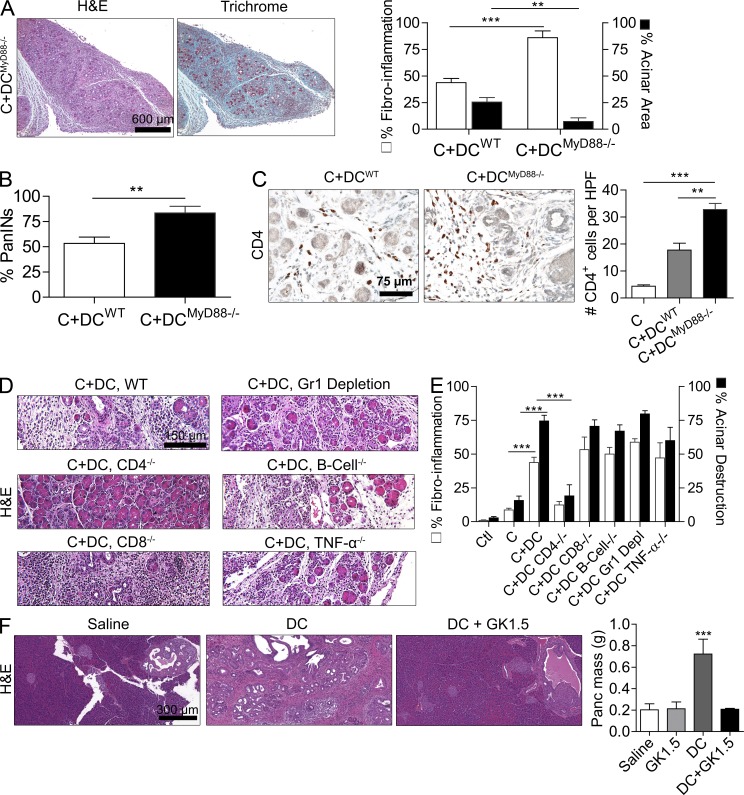
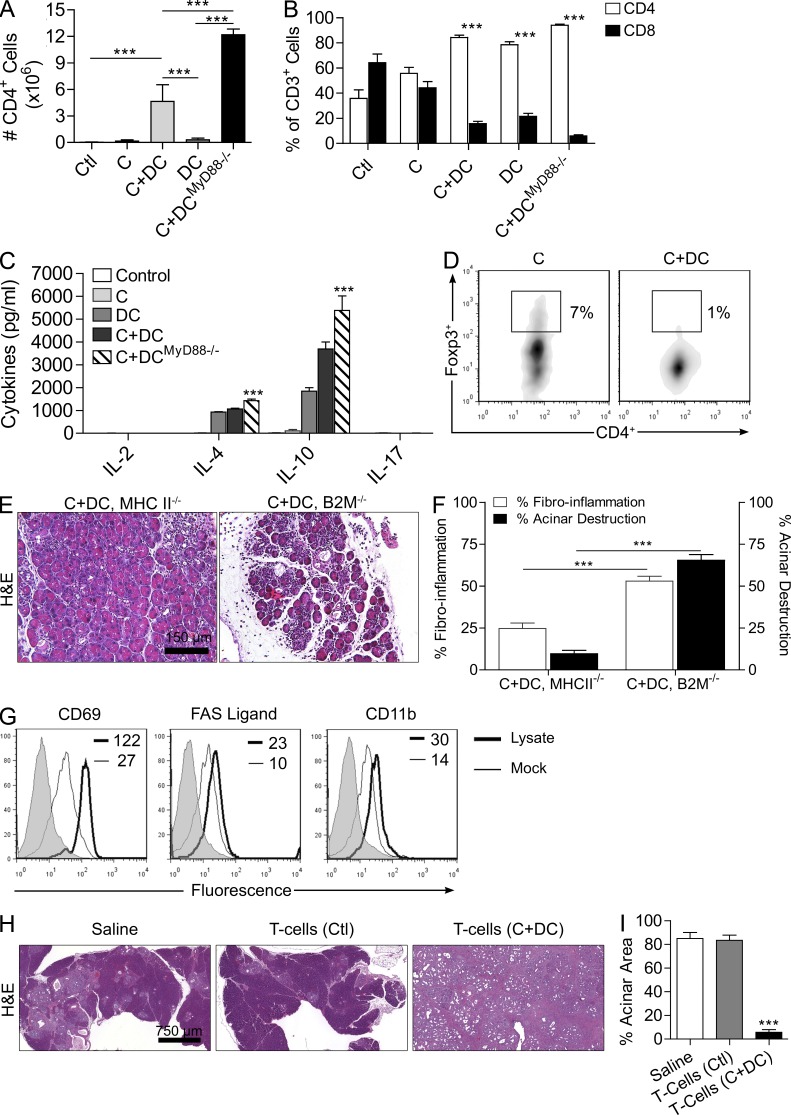
The transition of chronic pancreatic fibroinflammatory disease to neoplasia is a primary example of the paradigm linking inflammation to carcinogenesis. However, the cellular and molecular mediators bridging these entities are not well understood. Because TLR4 ligation can exacerbate pancreatic inflammation, we postulated that TLR4 activation drives pancreatic carcinogenesis. In this study, we show that lipopolysaccharide accelerates pancreatic tumorigenesis, whereas TLR4 inhibition is protective. Furthermore, blockade of the MyD88-independent TRIF pathway is protective against pancreatic cancer, whereas blockade of the MyD88-dependent pathway surprisingly exacerbates pancreatic inflammation and malignant progression. The protumorigenic and fibroinflammatory effects of MyD88 inhibition are mediated by dendritic cells (DCs), which induce pancreatic antigen-restricted Th2-deviated CD4(+) T cells and promote the transition from pancreatitis to carcinoma. Our data implicate a primary role for DCs in pancreatic carcinogenesis and illustrate divergent pathways in which blockade of TLR4 signaling via TRIF is protective against pancreatic cancer and, conversely, MyD88 inhibition exacerbates pancreatic inflammation and neoplastic transformation by augmenting the DC-Th2 axis.
Figures
Comment in
-
Tumour immunology: Differing roles for MYD88 in carcinogenesis.Nat Rev Immunol. 2012 Oct;12(10):681. doi: 10.1038/nri3304. Epub 2012 Sep 7. Nat Rev Immunol. 2012. PMID: 22955844 No abstract available.
References
-
- Ammann R.W., Akovbiantz A., Largiader F., Schueler G. 1984. Course and outcome of chronic pancreatitis. Longitudinal study of a mixed medical-surgical series of 245 patients. Gastroenterology. 86:820–828 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
