

Al amyloidosis
- PMID: 22909024
- PMCID: PMC3495844
- DOI: 10.1186/1750-1172-7-54
Al amyloidosis
Abstract
DEFINITION OF THE DISEASE: AL amyloidosis results from extra-cellular deposition of fibril-forming monoclonal immunoglobulin (Ig) light chains (LC) (most commonly of lambda isotype) usually secreted by a small plasma cell clone. Most patients have evidence of isolated monoclonal gammopathy or smoldering myeloma, and the occurrence of AL amyloidosis in patients with symptomatic multiple myeloma or other B-cell lymphoproliferative disorders is unusual. The key event in the development of AL amyloidosis is the change in the secondary or tertiary structure of an abnormal monoclonal LC, which results in instable conformation. This conformational change is responsible for abnormal folding of the LC, rich in β leaves, which assemble into monomers that stack together to form amyloid fibrils.
Epidemiology: AL amyloidosis is the most common type of systemic amyloidois in developed countries with an estimated incidence of 9 cases/million inhabitant/year. The average age of diagnosed patients is 65 years and less than 10% of patients are under 50.
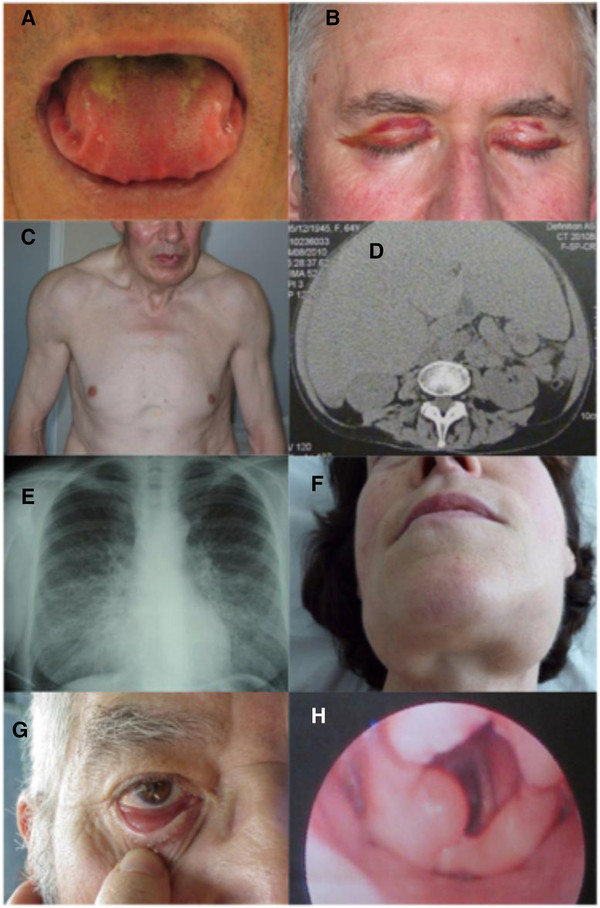
Clinical description: The clinical presentation is protean, because of the wide number of tissues or organs that may be affected. The most common presenting symptoms are asthenia and dyspnoea, which are poorly specific and may account for delayed diagnosis. Renal manifestations are the most frequent, affecting two thirds of patients at presentation. They are characterized by heavy proteinuria, with nephrotic syndrome and impaired renal function in half of the patients. Heart involvement, which is present at diagnosis in more than 50% of patients, leading to restrictive cardiopathy, is the most serious complication and engages prognosis.
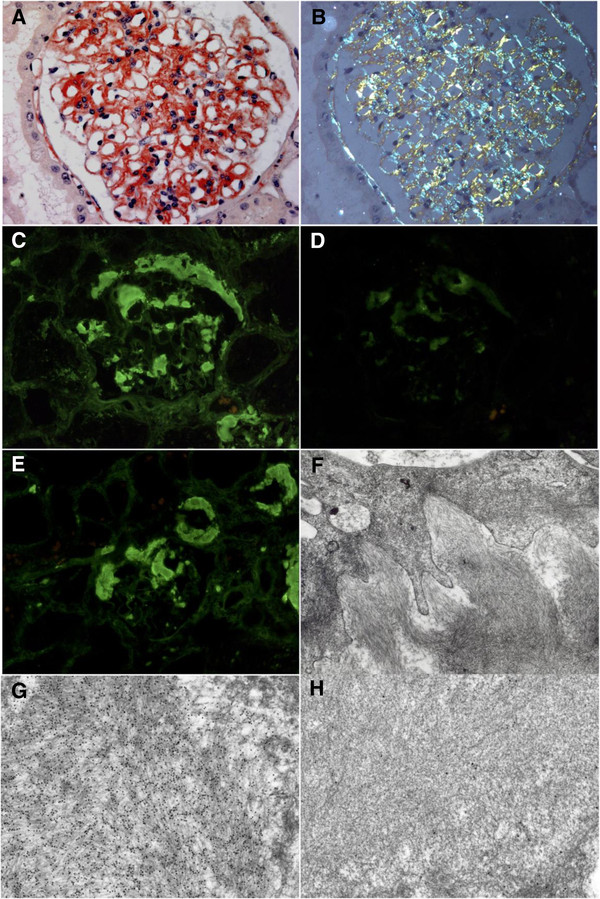
Diagnostic methods: The diagnosis relies on pathological examination of an involved site showing Congo red-positive amyloid deposits, with typical apple-green birefringence under polarized light, that stain positive with an anti-LC antibody by immunohistochemistry and/or immunofluorescence. Due to the systemic nature of the disease, non-invasive biopsies such as abdominal fat aspiration should be considered before taking biopsies from involved organs, in order to reduce the risk of bleeding complications.
Differential diagnosis: Systemic AL amyloidosis should be distinguished from other diseases related to deposition of monoclonal LC, and from other forms of systemic amyloidosis. When pathological studies have failed to identify the nature of amyloid deposits, genetic studies should be performed to diagnose hereditary amyloidosis.
Management: Treatment of AL amyloidosis is based on chemotherapy, aimed at controlling the underlying plasma clone that produces amyloidogenic LC. The hematological response should be carefully checked by serial measurements of serum free LC. The association of an alkylating agent with high-dose dexamethasone has proven to be effective in two thirds of patients and is considered as the current reference treatment. New agents used in the treatment of multiple myeloma are under investigation and appear to increase hematological response rates. Symptomatic measures and supportive care is necessary in patients with organ failure. Noticeably, usual treatments for cardiac failure (i.e. calcium inhibitors, β-blockers, angiotensin converting enzyme inhibitors) are inefficient or even dangerous in patients with amyloid heart disease, that should be managed using diuretics. Amiodarone and pace maker implantation should be considered in patients with rhythm or conduction abnormalities. In selected cases, heart and kidney transplantation may be associated with prolonged patient and graft survival.
Prognosis: Survival in AL amyloidosis depends on the spectrum of organ involvement (amyloid heart disease being the main prognosis factor), the severity of individual organs involved and haematological response to treatment.
Figures
References
-
- Kyle RA, Linos A, Beard CM, Linke RP, Gertz MA, O’Fallon WM, Kurland LT. Incidence and natural history of primary systemic amyloidosis in Olmstead County, Minesota, 1950 through 1989. Blood. 1992;79:1817–1822. - PubMed
-
- Gertz MA, Comenzo R, Falk RH, Fermand JP, Hazenberg BP, Hawkins PN, Merlini G, Moreau P, Ronco P, Sanchorawala V, Sezer O, Solomon A, Grateau G. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis, Tours, France, 18–22 April 2004. Am J Hematol. 2005;79:319–328. doi: 10.1002/ajh.20381. - DOI - PubMed
-
- Gertz MA, Merlini G. Definition of organ involvement and response to treatment in AL amyloidosis: an updated consensus opinion. Amyloid. 2010;17(Suppl 1):48–49.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
