

AATF/Che-1 acts as a phosphorylation-dependent molecular modulator to repress p53-driven apoptosis
- PMID: 22909821
- PMCID: PMC3474921
- DOI: 10.1038/emboj.2012.236
AATF/Che-1 acts as a phosphorylation-dependent molecular modulator to repress p53-driven apoptosis
Abstract
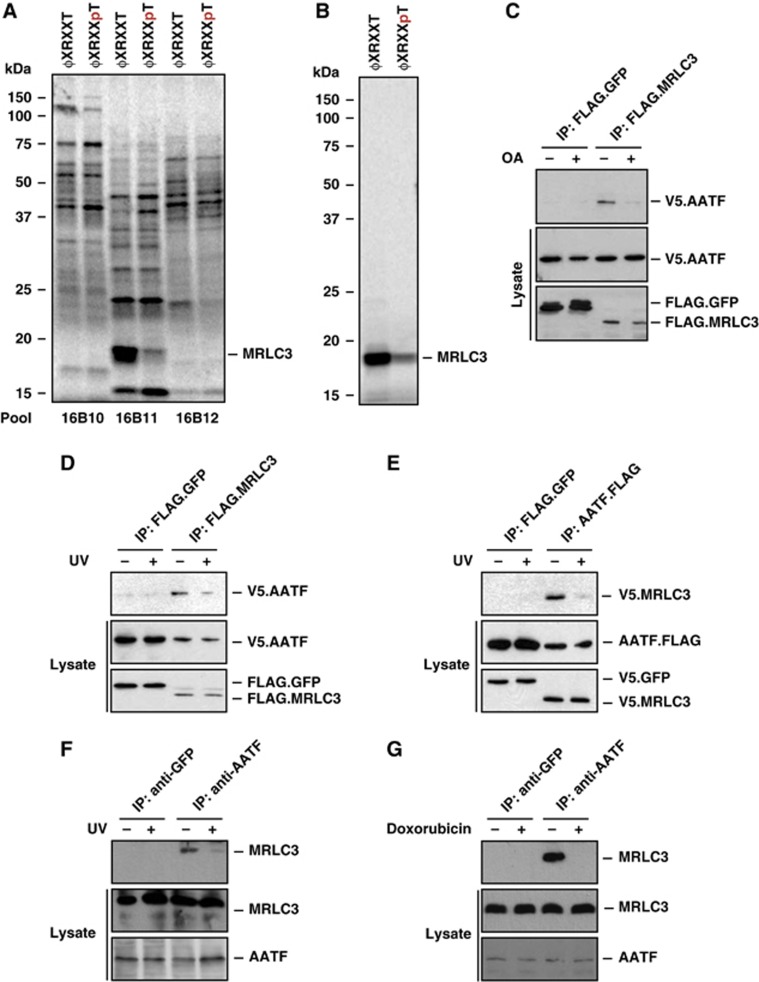
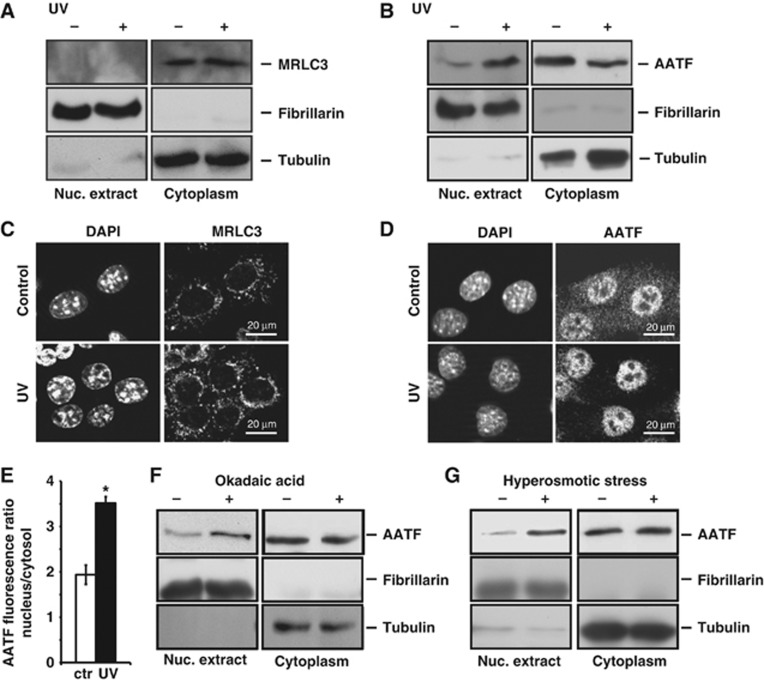
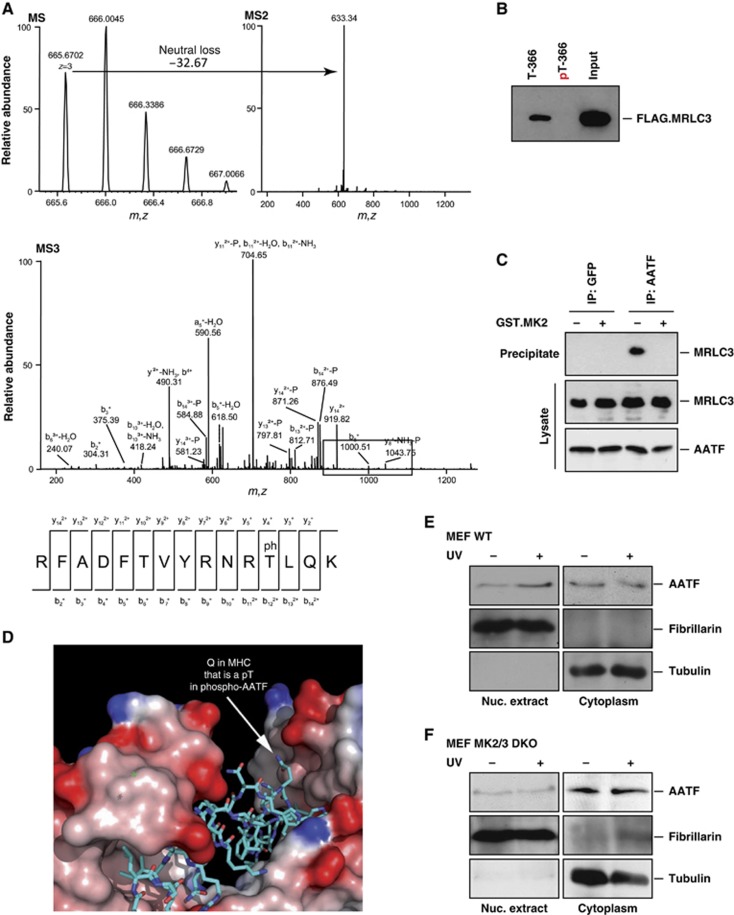
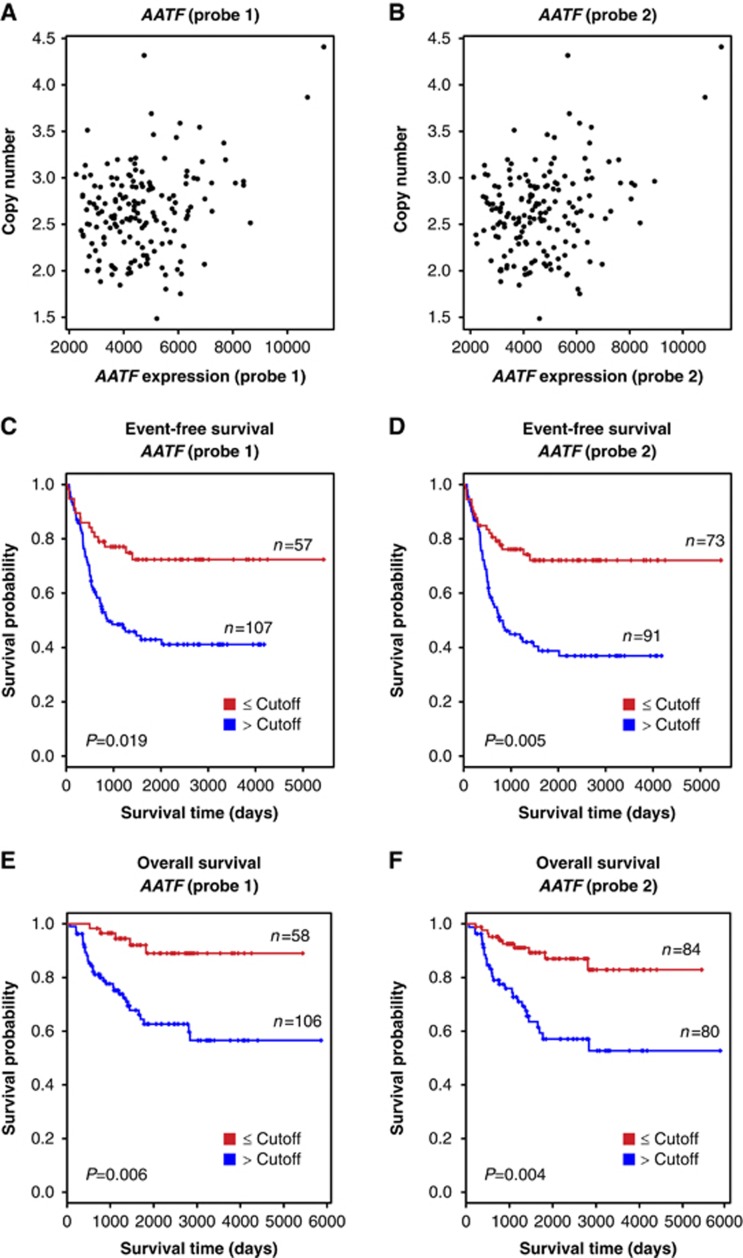
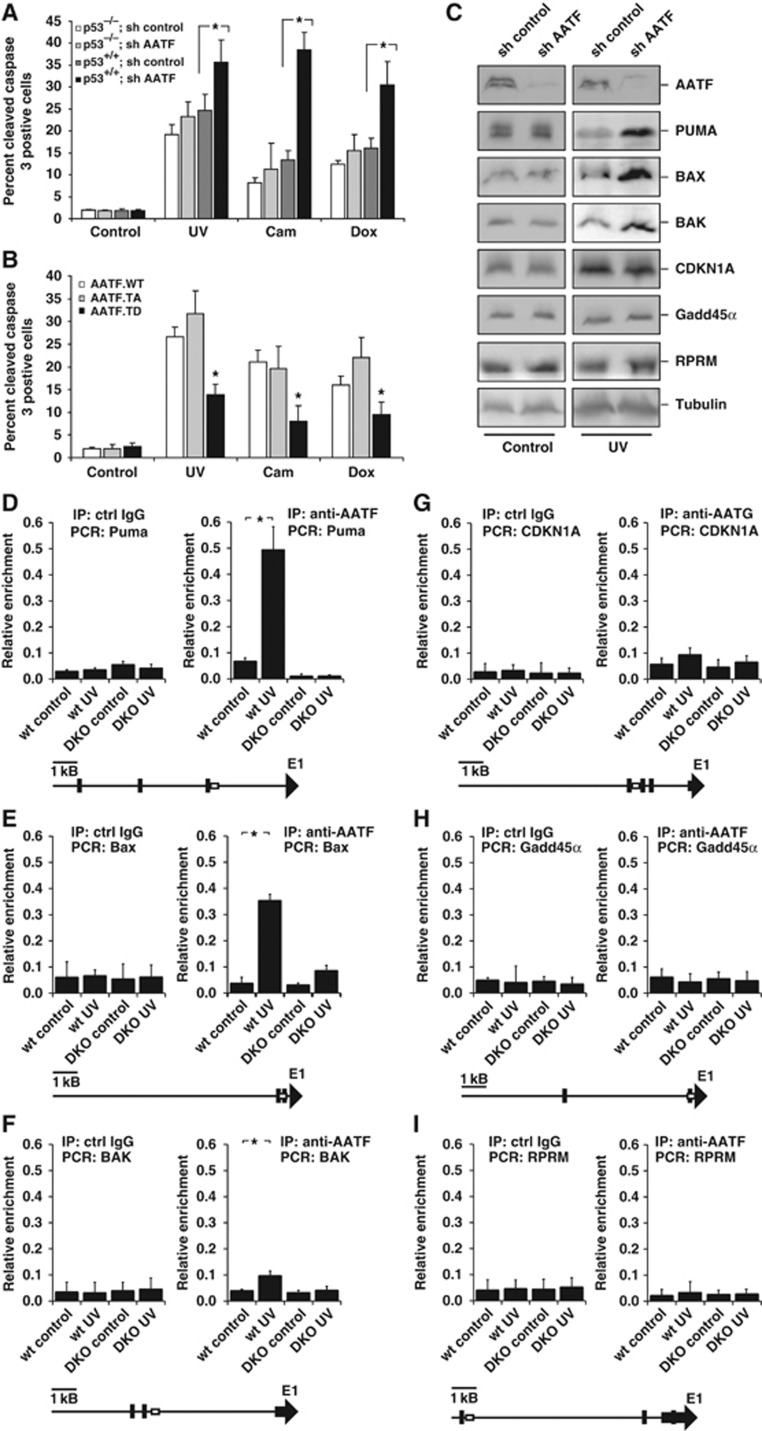
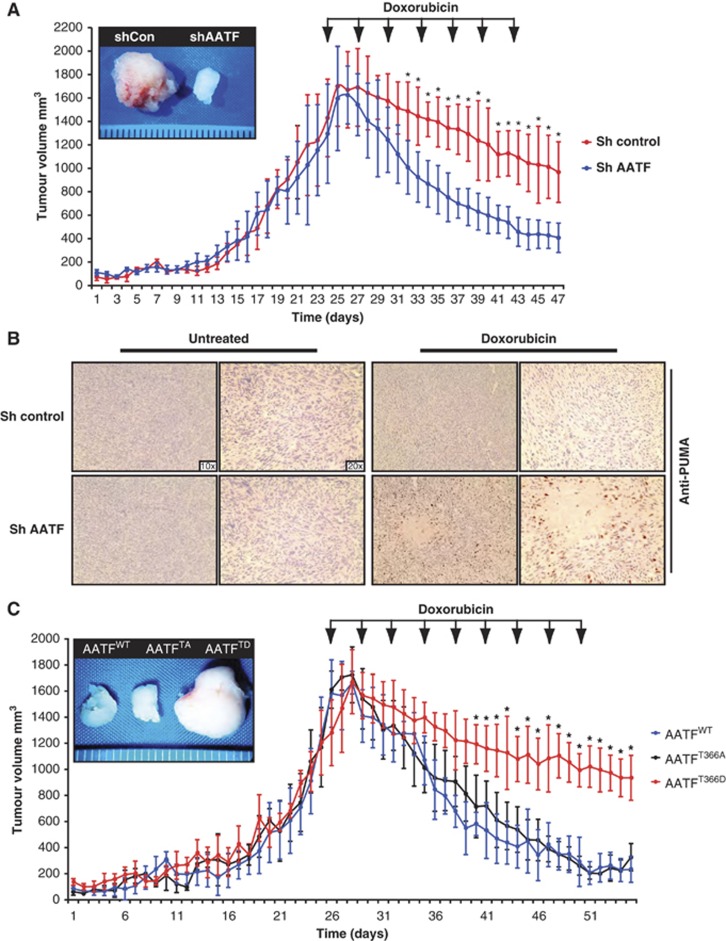
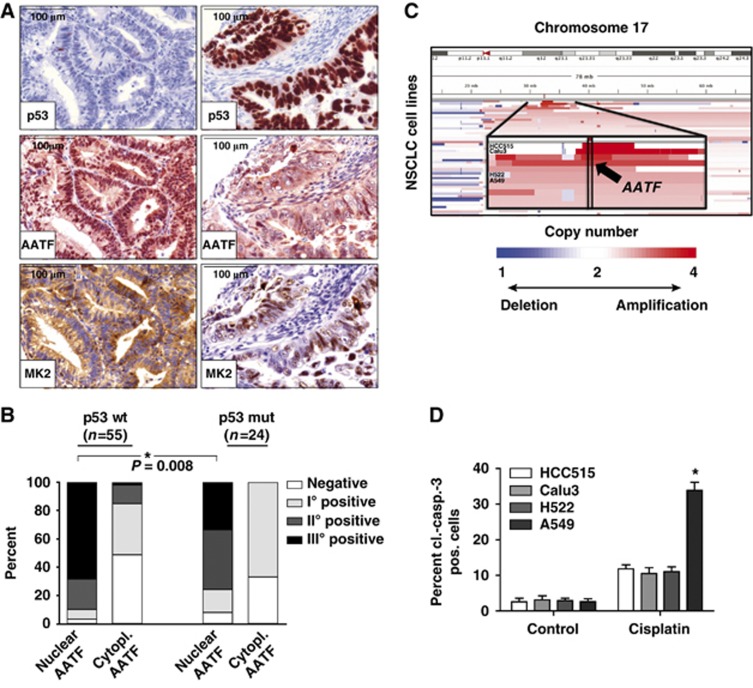
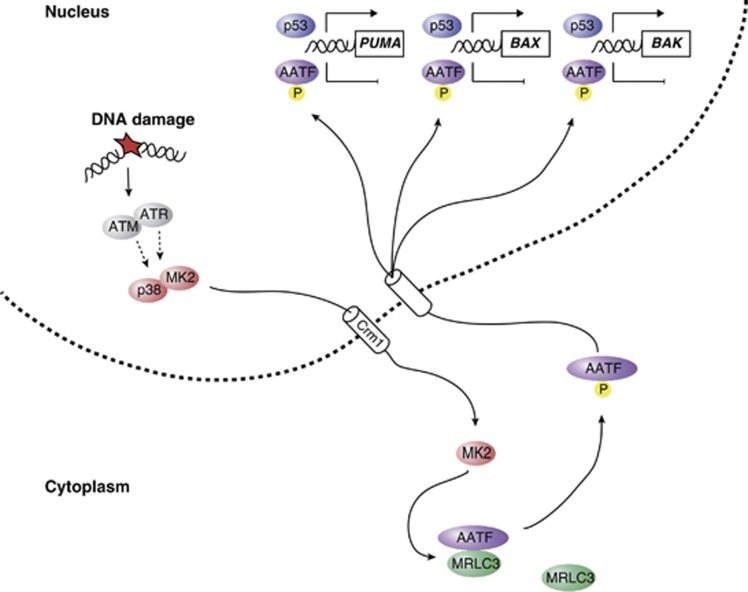
Following genotoxic stress, cells activate a complex signalling network to arrest the cell cycle and initiate DNA repair or apoptosis. The tumour suppressor p53 lies at the heart of this DNA damage response. However, it remains incompletely understood, which signalling molecules dictate the choice between these different cellular outcomes. Here, we identify the transcriptional regulator apoptosis-antagonizing transcription factor (AATF)/Che-1 as a critical regulator of the cellular outcome of the p53 response. Upon genotoxic stress, AATF is phosphorylated by the checkpoint kinase MK2. Phosphorylation results in the release of AATF from cytoplasmic MRLC3 and subsequent nuclear translocation where AATF binds to the PUMA, BAX and BAK promoter regions to repress p53-driven expression of these pro-apoptotic genes. In xenograft experiments, mice exhibit a dramatically enhanced response of AATF-depleted tumours following genotoxic chemotherapy with adriamycin. The exogenous expression of a phospho-mimicking AATF point mutant results in marked adriamycin resistance in vivo. Nuclear AATF enrichment appears to be selected for in p53-proficient endometrial cancers. Furthermore, focal copy number gains at the AATF locus in neuroblastoma, which is known to be almost exclusively p53-proficient, correlate with an adverse prognosis and reduced overall survival. These data identify the p38/MK2/AATF signalling module as a critical repressor of p53-driven apoptosis and commend this pathway as a target for DNA damage-sensitizing therapeutic regimens.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
Comment in
-
Che-ating death: CHE1/AATF protects from p53-mediated apoptosis.EMBO J. 2012 Oct 17;31(20):3951-3. doi: 10.1038/emboj.2012.258. Epub 2012 Sep 7. EMBO J. 2012. PMID: 22960635 Free PMC article.
References
-
- Bruno T, De Angelis R, De Nicola F, Barbato C, Di Padova M, Corbi N, Libri V, Benassi B, Mattei E, Chersi A, Soddu S, Floridi A, Passananti C, Fanciulli M (2002) Che-1 affects cell growth by interfering with the recruitment of HDAC1 by Rb. Cancer Cell 2: 387–399 - PubMed
-
- Bruno T, De Nicola F, Iezzi S, Lecis D, D'Angelo C, Di Padova M, Corbi N, Dimiziani L, Zannini L, Jekimovs C, Scarsella M, Porrello A, Chersi A, Crescenzi M, Leonetti C, Khanna KK, Soddu S, Floridi A, Passananti C, Delia D et al. (2006) Che-1 phosphorylation by ATM/ATR and Chk2 kinases activates p53 transcription and the G2/M checkpoint. Cancer Cell 10: 473–486 - PubMed
-
- Bruno T, Iezzi S, De Nicola F, Di Padova M, Desantis A, Scarsella M, Di Certo MG, Leonetti C, Floridi A, Passananti C, Fanciulli M (2008) Che-1 activates XIAP expression in response to DNA damage. Cell Death Differ 15: 515–520 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
