

The peroxisome proliferator-activated receptor γ (PPARγ) controls natural protective mechanisms against lipid peroxidation in amyotrophic lateral sclerosis
- PMID: 22910911
- PMCID: PMC3476258
- DOI: 10.1074/jbc.M112.366419
The peroxisome proliferator-activated receptor γ (PPARγ) controls natural protective mechanisms against lipid peroxidation in amyotrophic lateral sclerosis
Abstract
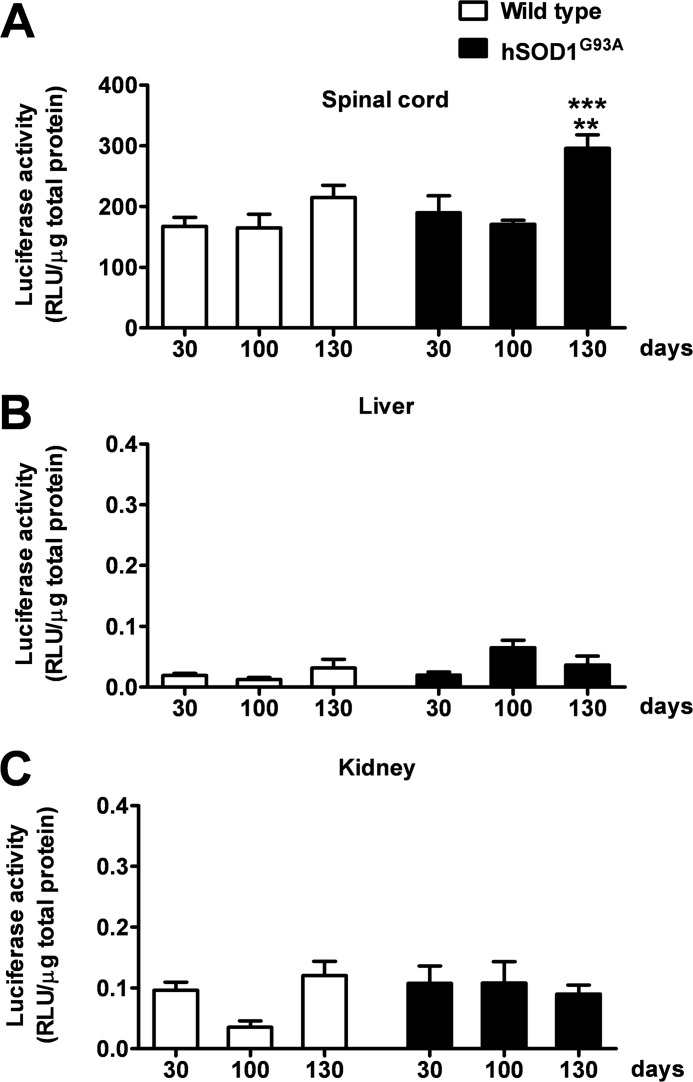
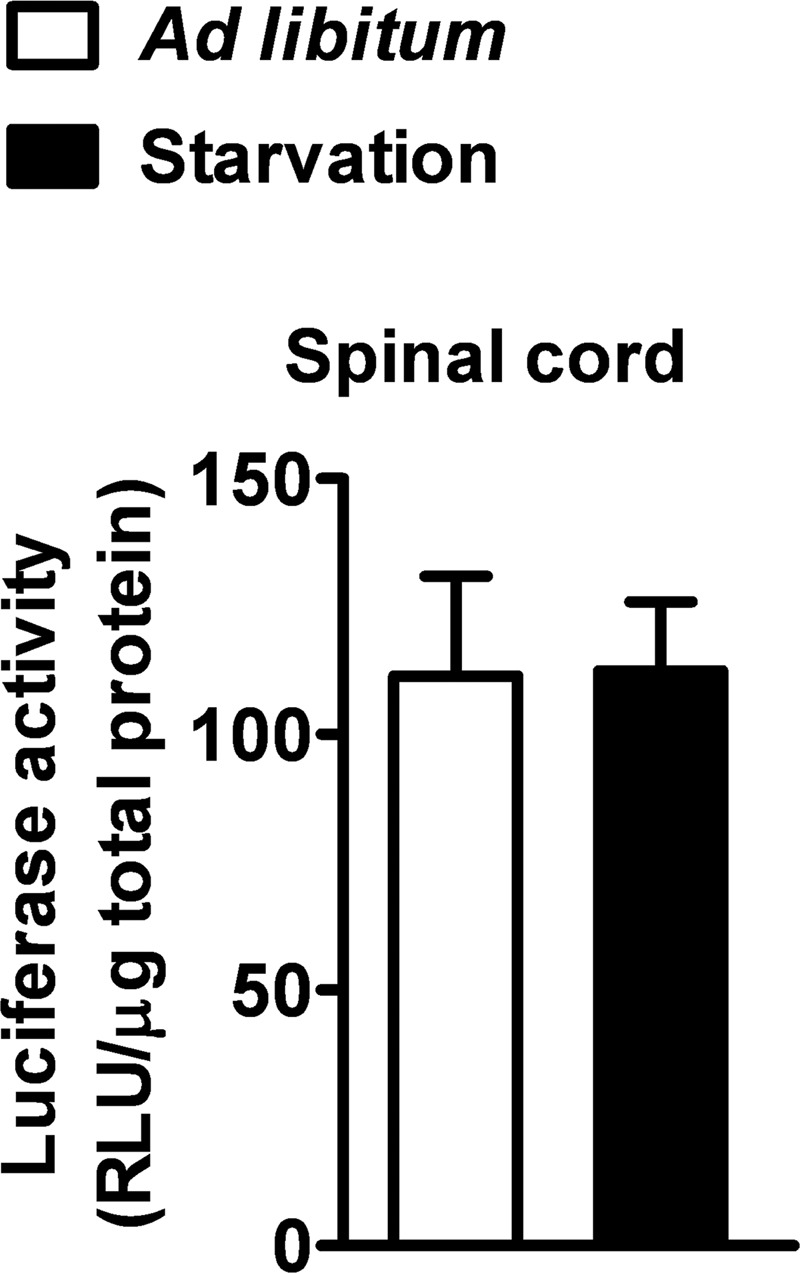
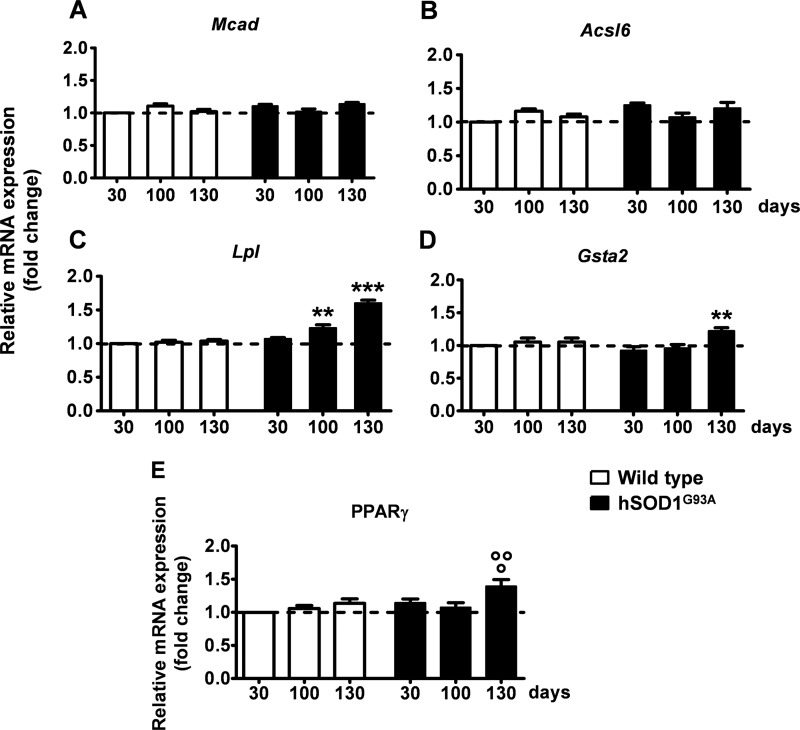
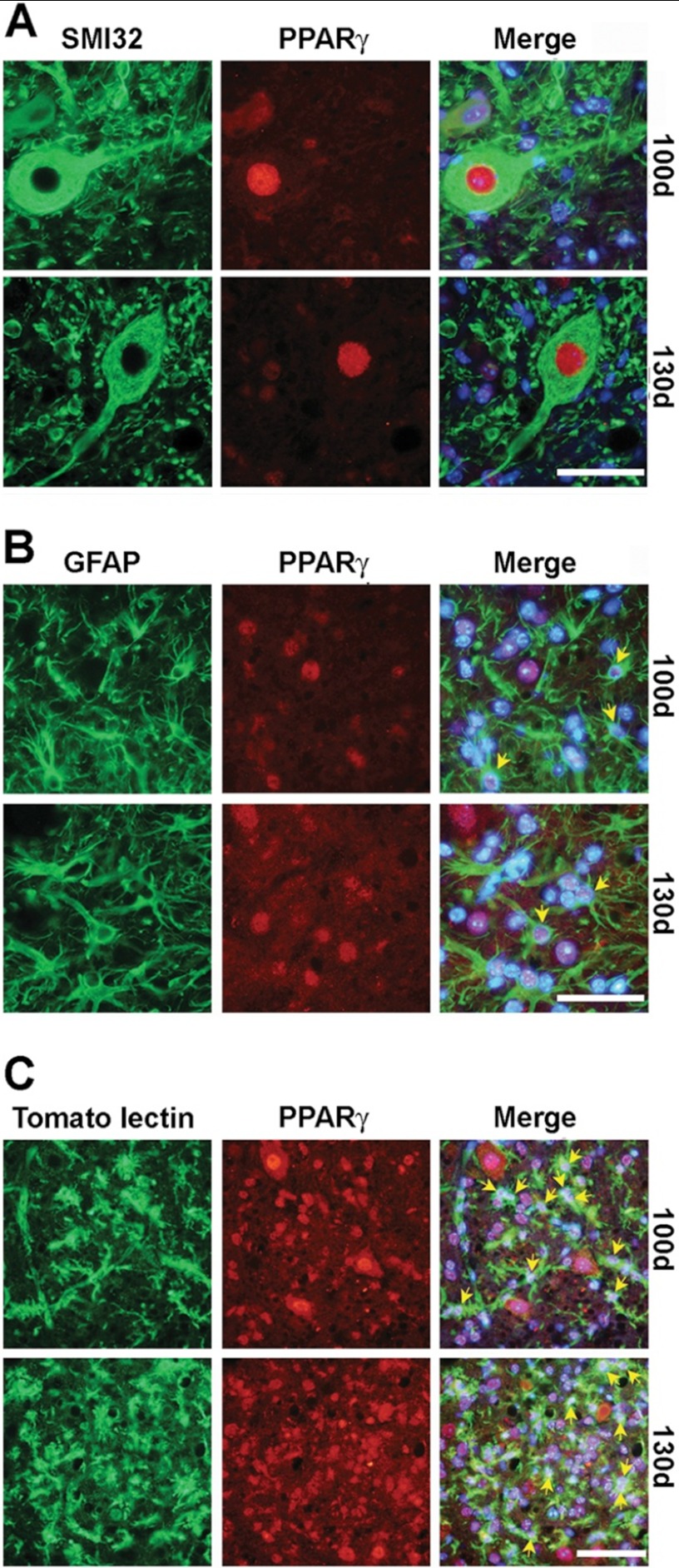
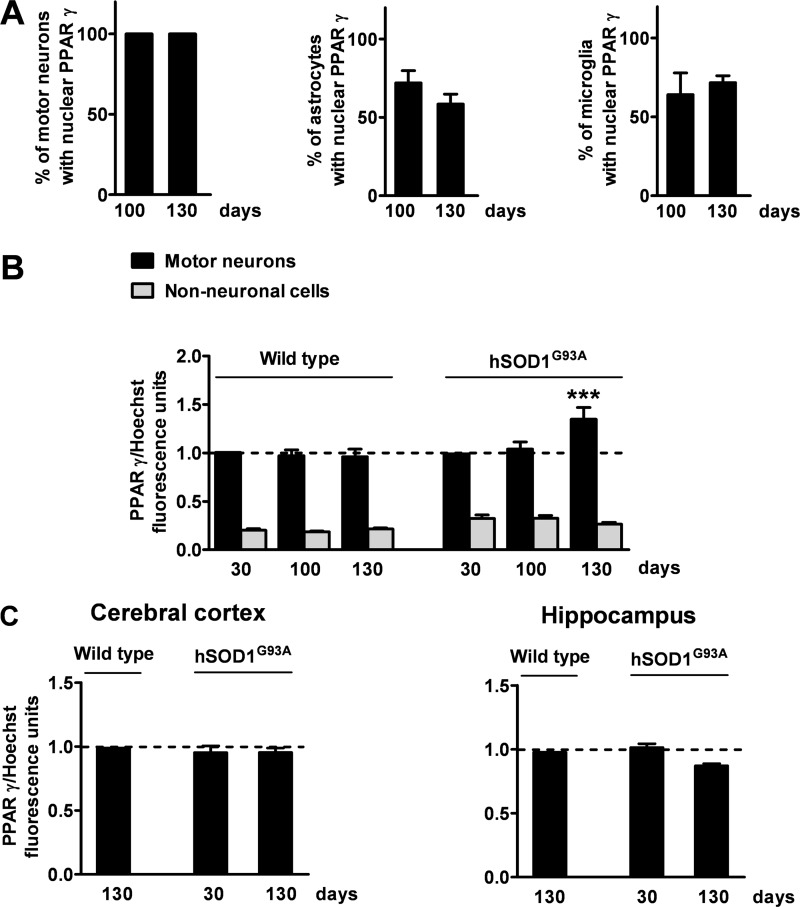
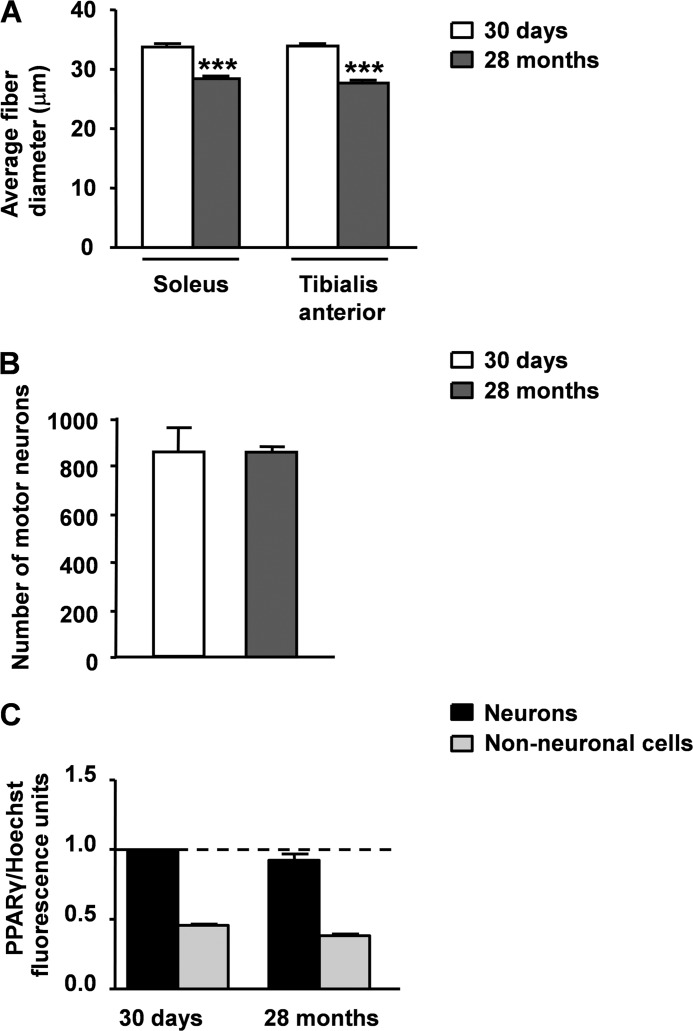
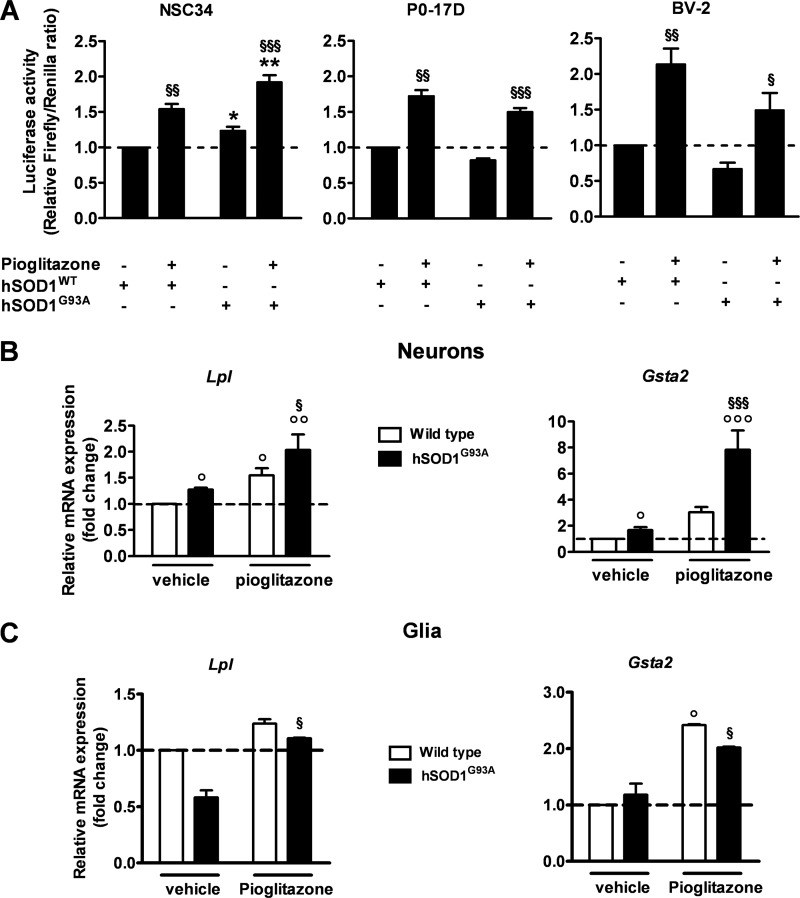
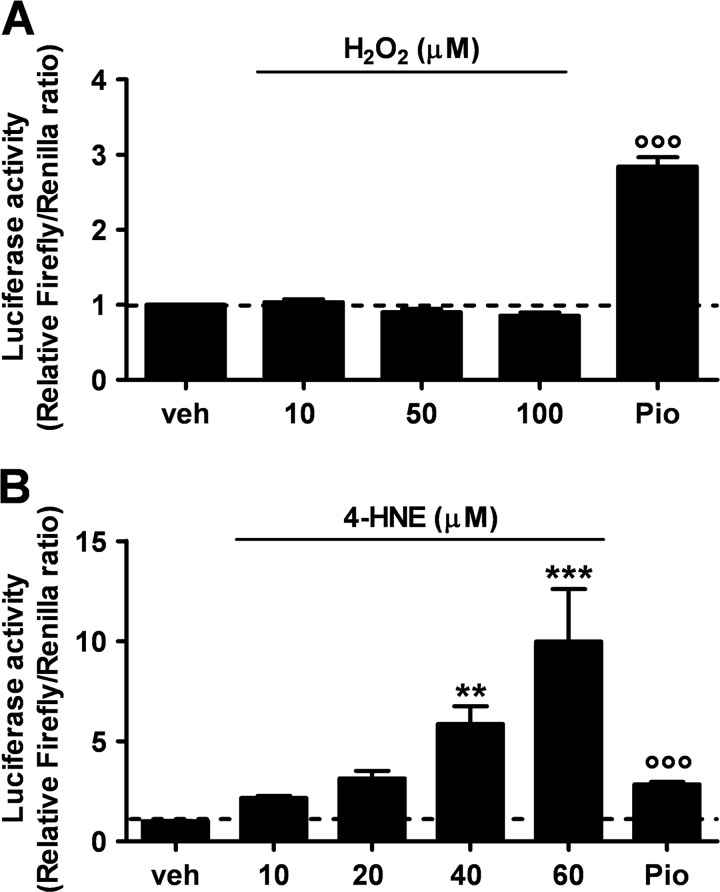
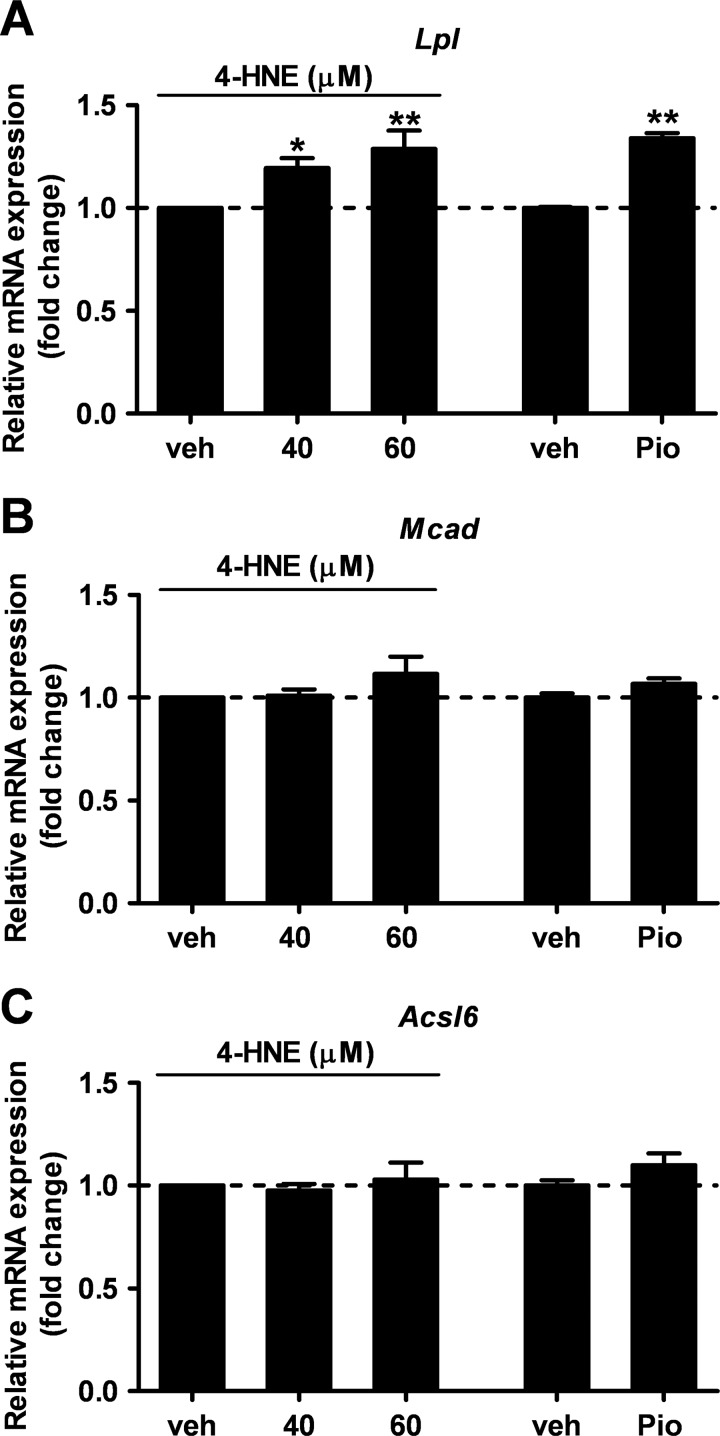
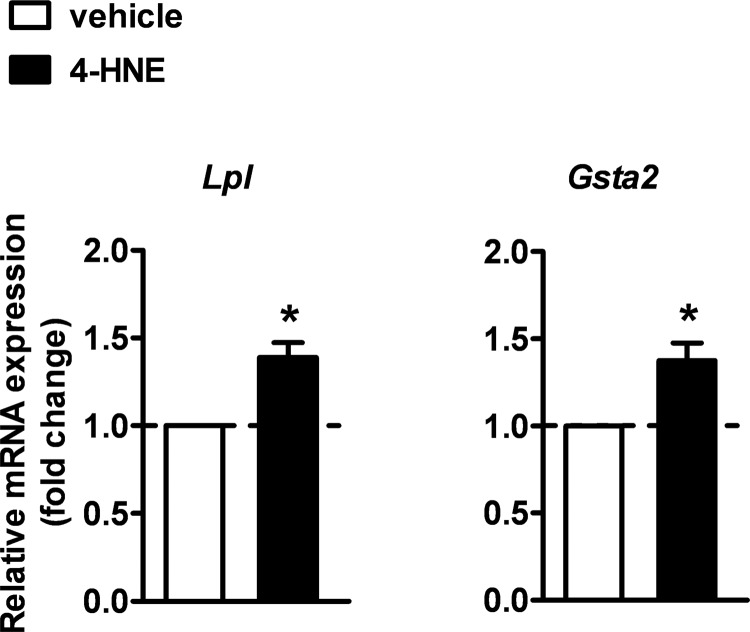
Recent evidence highlights the peroxisome proliferator-activated receptors (PPARs) as critical neuroprotective factors in several neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS). To gain new mechanistic insights into the role of these receptors in the context of ALS, here we investigated how PPAR transcriptional activity varies in hSOD1(G93A) ALS transgenic mice. We demonstrate that PPARγ-driven transcription selectively increases in the spinal cord of symptomatic hSOD1(G93A) mice. This phenomenon correlates with the up-regulation of target genes, such as lipoprotein lipase and glutathione S-transferase α-2, which are implicated in scavenging lipid peroxidation by-products. Such events are associated with enhanced PPARγ immunoreactivity within motor neuronal nuclei. This observation, and the fact that PPARγ displays increased responsiveness in cultured hSOD1(G93A) motor neurons, points to a role for this receptor in neutralizing deleterious lipoperoxidation derivatives within the motor cells. Consistently, in both motor neuron-like cultures and animal models, we report that PPARγ is activated by lipid peroxidation end products, such as 4-hydroxynonenal, whose levels are elevated in the cerebrospinal fluid and spinal cord from ALS patients. We propose that the accumulation of critical concentrations of lipid peroxidation adducts during ALS progression leads to the activation of PPARγ in motor neurons. This in turn triggers self-protective mechanisms that involve the up-regulation of lipid detoxification enzymes, such as lipoprotein lipase and glutathione S-transferase α-2. Our findings indicate that anticipating natural protective reactions by pharmacologically modulating PPARγ transcriptional activity may attenuate neurodegeneration by limiting the damage induced by lipid peroxidation derivatives.
Figures
References
-
- Rosen D. R., Siddique T., Patterson D., Figlewicz D. A., Sapp P., Hentati A., Donaldson D., Goto J., O'Regan J. P., Deng H. X. (1993) Mutations in Cu/Zn-superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62 - PubMed
-
- Gurney M. E., Pu H., Chiu A. Y., Dal Canto M. C., Polchow C. Y., Alexander D. D., Caliendo J., Hentati A., Kwon Y. W., Deng H. X. (1994) Motor neuron degeneration in mice that express a human Cu,Zn-superoxide dismutase mutation. Science 264, 1772–1775 - PubMed
-
- Bruijn L. I., Becher M. W., Lee M. K., Anderson K. L., Jenkins N. A., Copeland N. G., Sisodia S. S., Rothstein J. D., Borchelt D. R., Price D. L., Cleveland D. W. (1997) ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 18, 327–338 - PubMed
-
- Wong P. C., Pardo C. A., Borchelt D. R., Lee M. K., Copeland N. G., Jenkins N. A., Sisodia S. S., Cleveland D. W., Price D. L. (1995) An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 14, 1105–1116 - PubMed
-
- Turner B. J., Talbot K. (2008) Transgenics, toxicity, and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog. Neurobiol. 85, 94–134 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
