

Type I interferons promote fatal immunopathology by regulating inflammatory monocytes and neutrophils during Candida infections
- PMID: 22911155
- PMCID: PMC3406095
- DOI: 10.1371/journal.ppat.1002811
Type I interferons promote fatal immunopathology by regulating inflammatory monocytes and neutrophils during Candida infections
Abstract
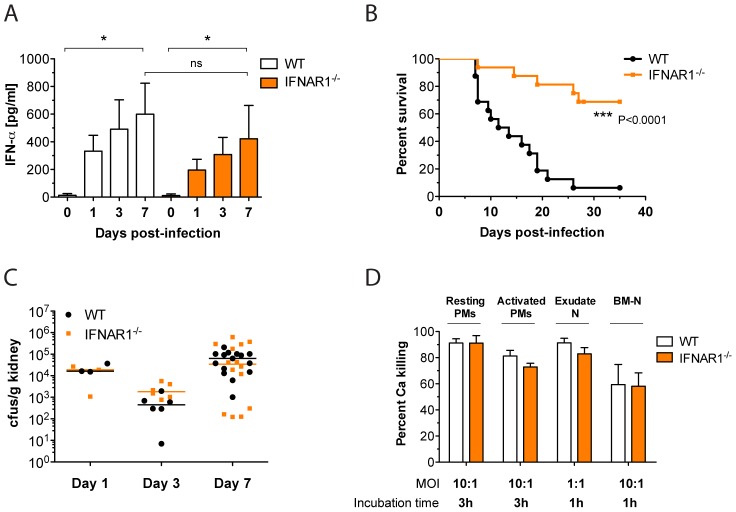
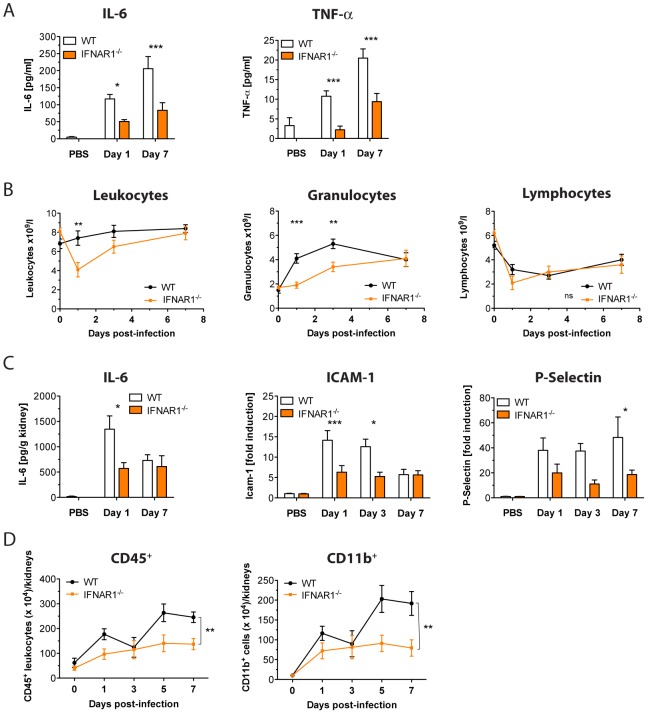
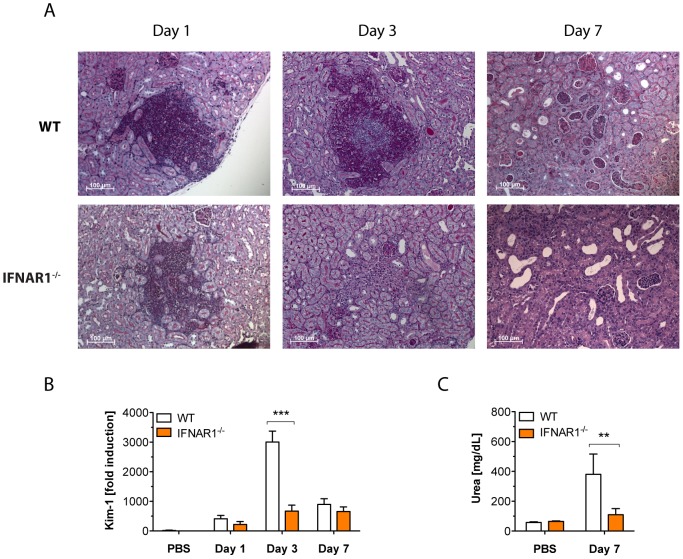
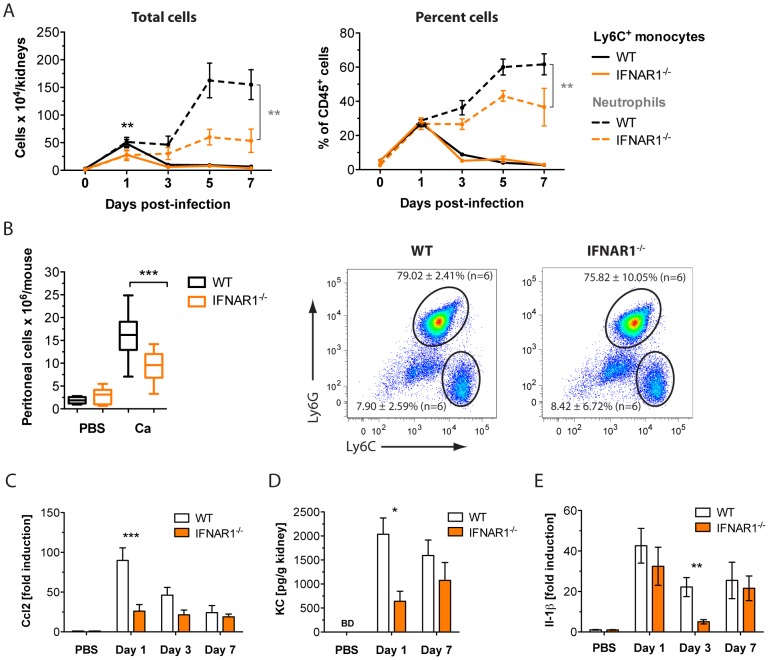
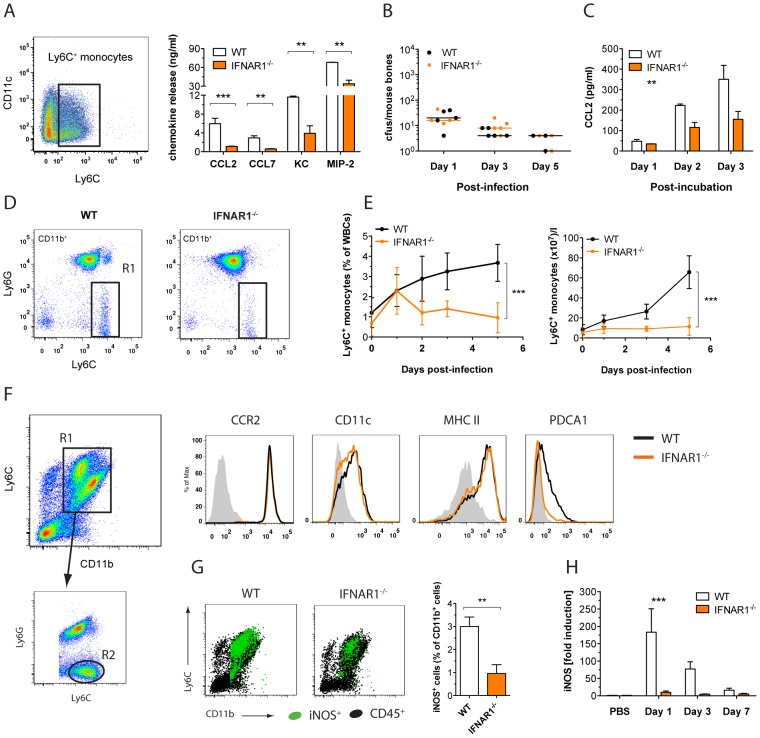
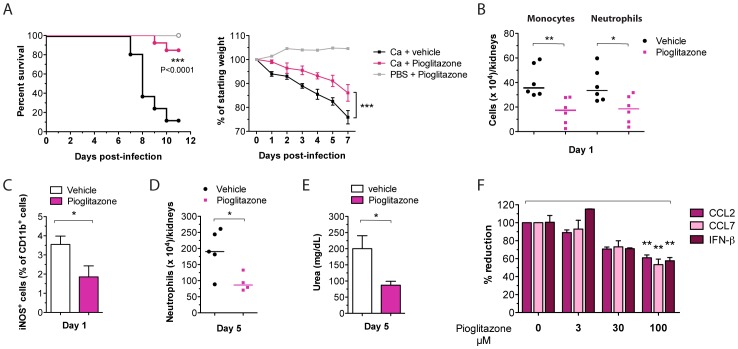
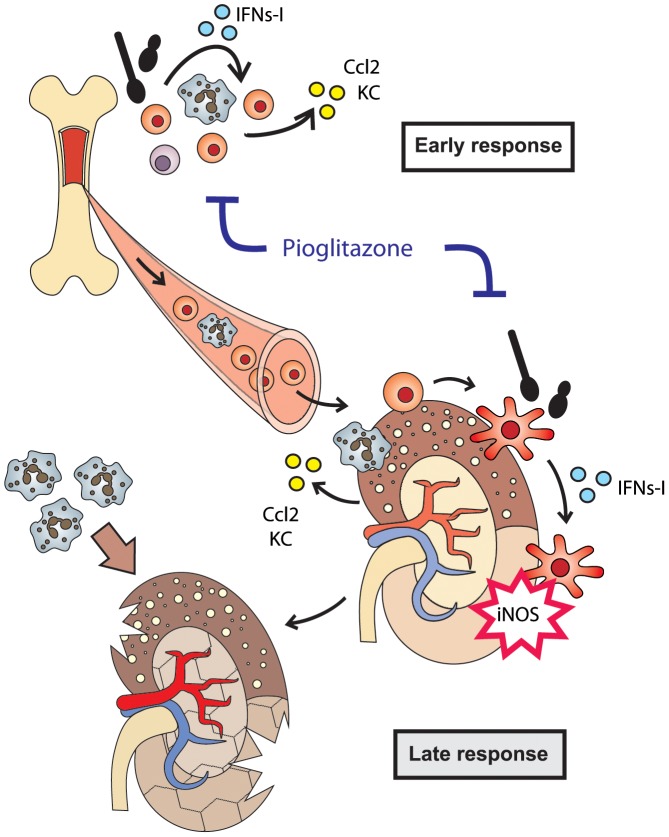
Invasive fungal infections by Candida albicans (Ca) are a frequent cause of lethal sepsis in intensive care unit patients. While a contribution of type I interferons (IFNs-I) in fungal sepsis remains unknown, these immunostimulatory cytokines mediate the lethal effects of endotoxemia and bacterial sepsis. Using a mouse model lacking a functional IFN-I receptor (Ifnar1⁻/⁻), we demonstrate a remarkable protection against invasive Ca infections. We discover a mechanism whereby IFN-I signaling controls the recruitment of inflammatory myeloid cells, including Ly6C(hi) monocytes and neutrophils, to infected kidneys by driving expression of the chemokines CCL2 and KC. Within kidneys, monocytes differentiate into inflammatory DCs but fail to functionally mature in Ifnar1⁻/⁻ mice, as demonstrated by the impaired upregulation of the key activation markers PDCA1 and iNOS. The increased activity of inflammatory monocytes and neutrophils results in hyper-inflammation and lethal kidney pathology. Pharmacological diminution of monocytes and neutrophils by treating mice with pioglitazone, a synthetic agonist of the nuclear receptor peroxisome proliferator-activated receptor-γ (PPAR-γ), strongly reduces renal immunopathology during Ca infection and improves mouse survival. Taken together, our data connect for the first time the sepsis-promoting functions of IFNs-I to the CCL2-mediated recruitment and the activation of inflammatory monocytes/DCs with high host-destructing potency. Moreover, our data demonstrate a therapeutic relevance of PPAR-γ agonists for microbial infectious diseases where inflammatory myeloid cells may contribute to fatal tissue damage.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Wisplinghoff H, Seifert H, Wenzel RP, Edmond MB. Inflammatory response and clinical course of adult patients with nosocomial bloodstream infections caused by Candida spp. Clin Microbiol Infect. 2006;12:170–177. - PubMed
-
- Decker T, Muller M, Stockinger S. The yin and yang of type I interferon activity in bacterial infection. Nat Rev Immunol. 2005;5:675–687. - PubMed
-
- Bourgeois C, Majer O, Frohner IE, Lesiak-Markowicz I, Hildering KS, et al. Conventional dendritic cells mount a type I IFN response against Candida spp. requiring novel phagosomal TLR7-mediated IFN-beta signaling. J Immunol. 2011;186:3104–3112. - PubMed
-
- Biondo C, Signorino G, Costa A, Midiri A, Gerace E, et al. Recognition of yeast nucleic acids triggers a host-protective type I interferon response. Eur J Immunol. 2011;41:1969–1979. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
