

Systematic dissection of roles for chromatin regulators in a yeast stress response
- PMID: 22912562
- PMCID: PMC3416867
- DOI: 10.1371/journal.pbio.1001369
Systematic dissection of roles for chromatin regulators in a yeast stress response
Abstract
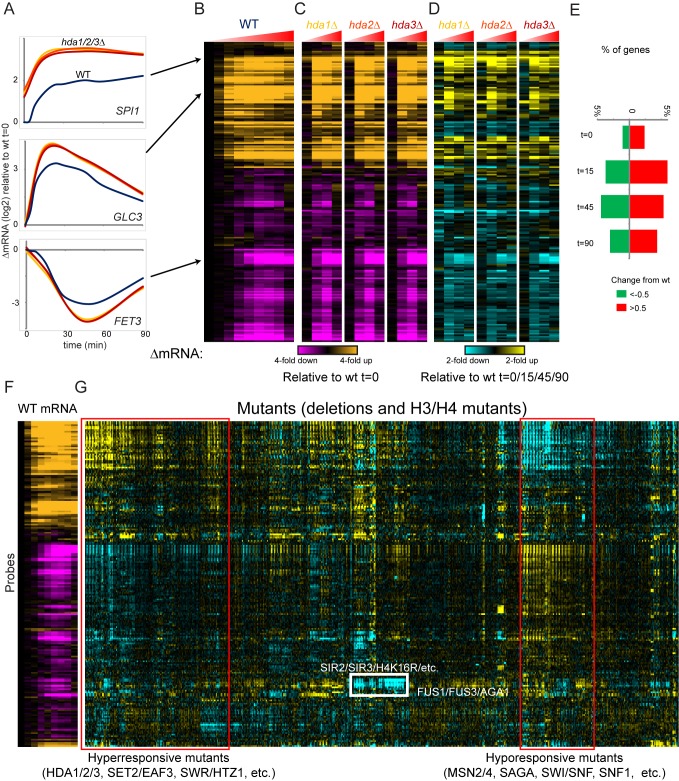
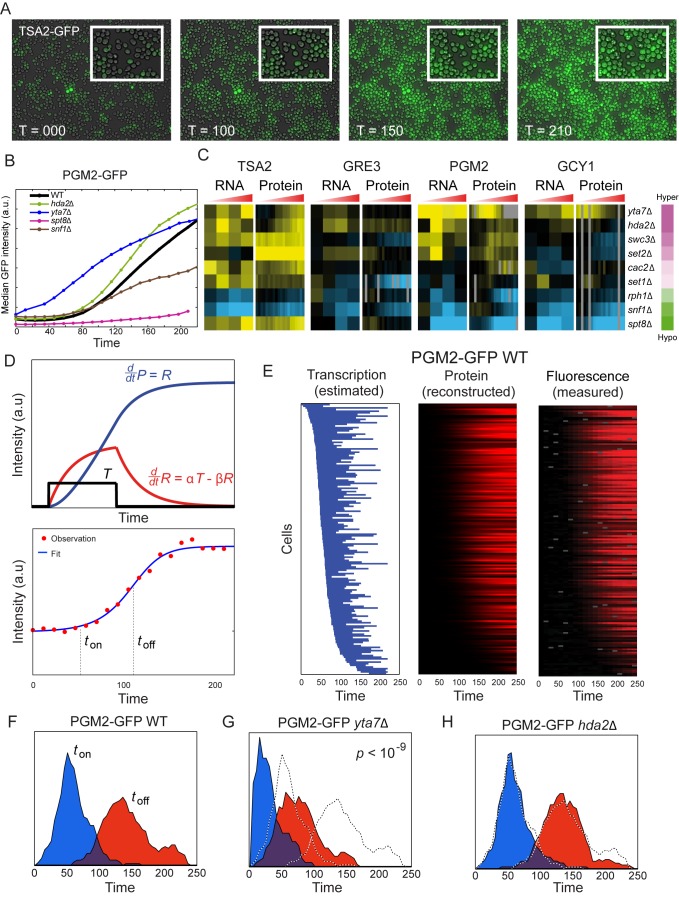
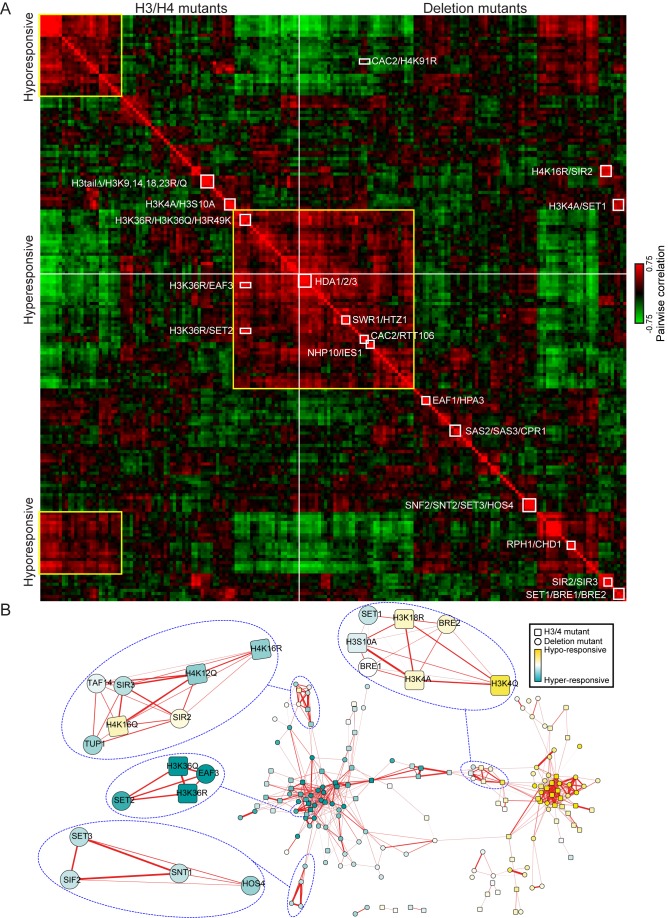
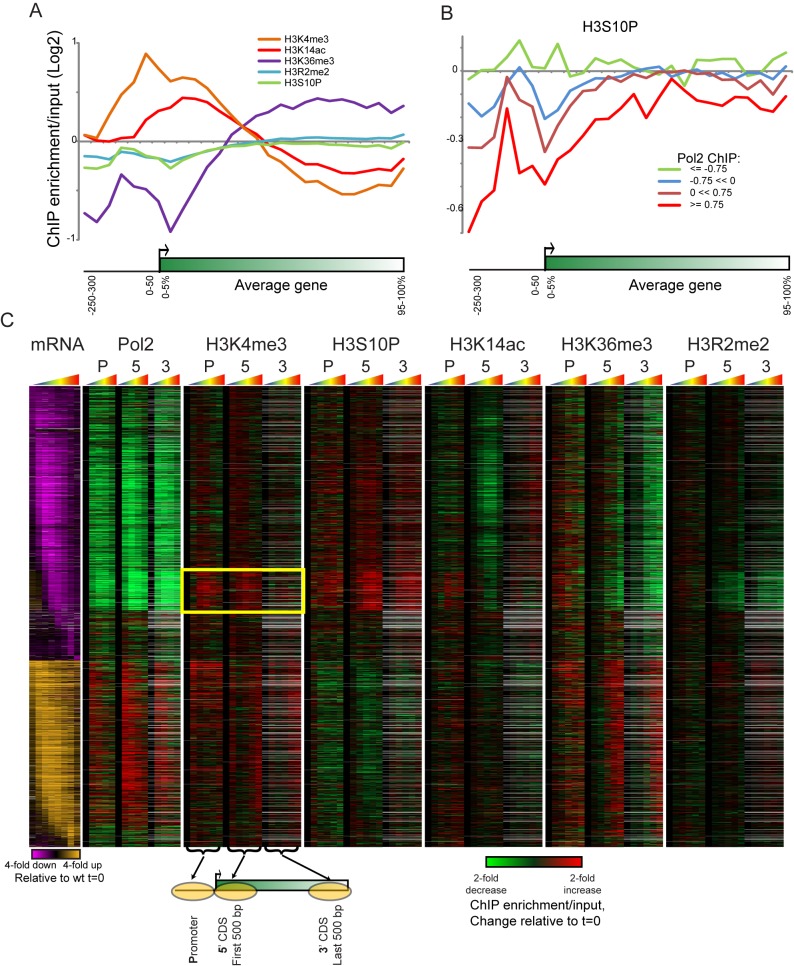
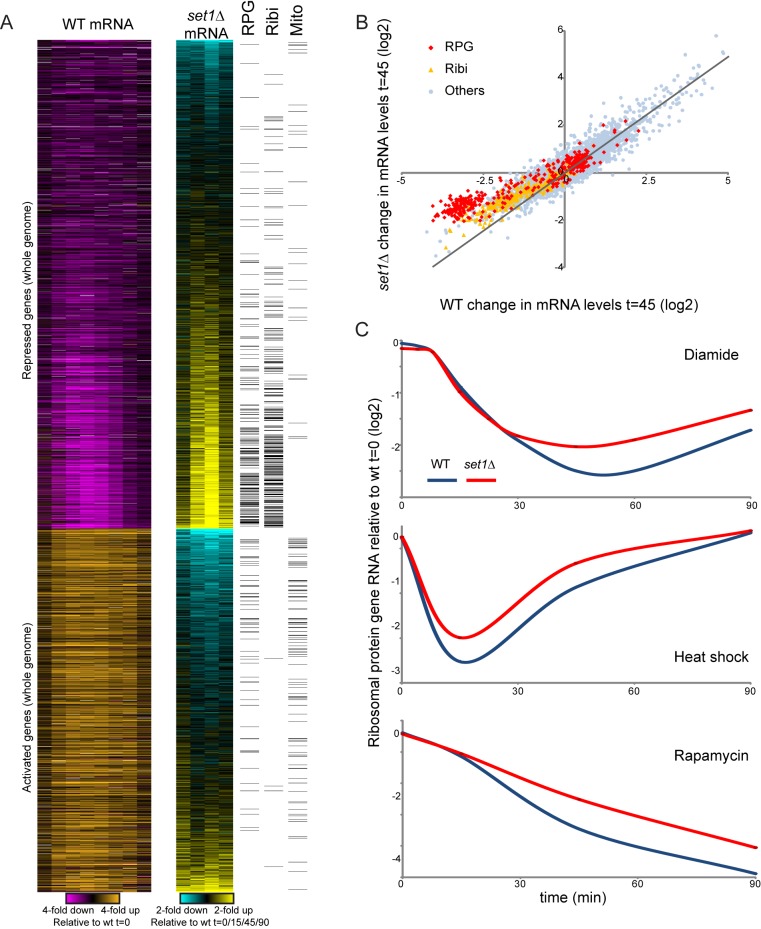
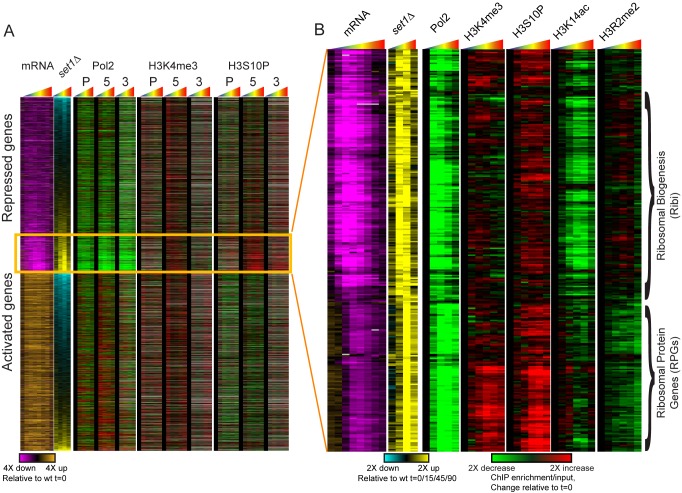
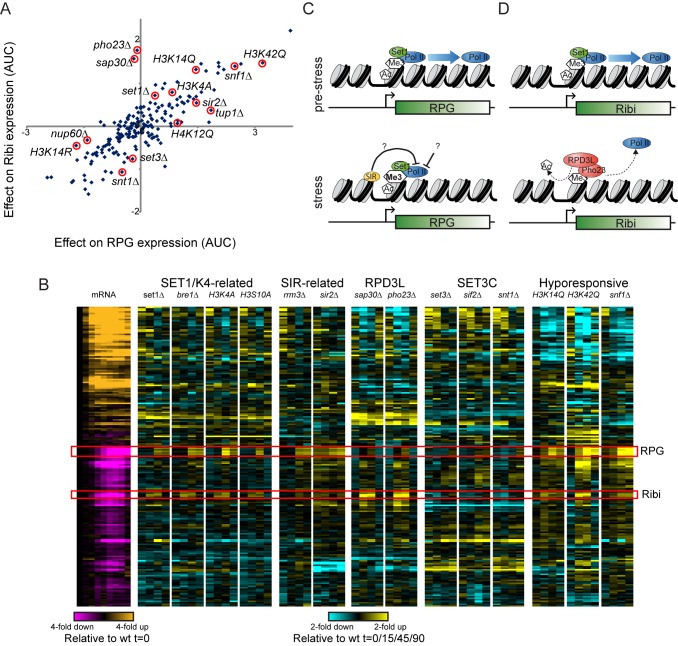
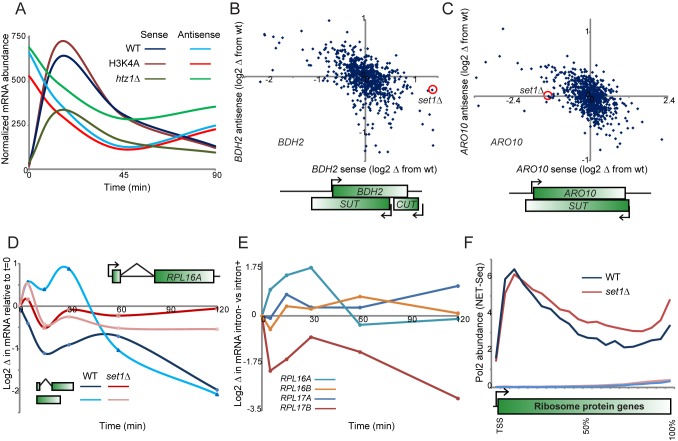
Packaging of eukaryotic genomes into chromatin has wide-ranging effects on gene transcription. Curiously, it is commonly observed that deletion of a global chromatin regulator affects expression of only a limited subset of genes bound to or modified by the regulator in question. However, in many single-gene studies it has become clear that chromatin regulators often do not affect steady-state transcription, but instead are required for normal transcriptional reprogramming by environmental cues. We therefore have systematically investigated the effects of 83 histone mutants, and 119 gene deletion mutants, on induction/repression dynamics of 170 transcripts in response to diamide stress in yeast. Importantly, we find that chromatin regulators play far more pronounced roles during gene induction/repression than they do in steady-state expression. Furthermore, by jointly analyzing the substrates (histone mutants) and enzymes (chromatin modifier deletions) we identify specific interactions between histone modifications and their regulators. Combining these functional results with genome-wide mapping of several histone marks in the same time course, we systematically investigated the correspondence between histone modification occurrence and function. We followed up on one pathway, finding that Set1-dependent H3K4 methylation primarily acts as a gene repressor during multiple stresses, specifically at genes involved in ribosome biosynthesis. Set1-dependent repression of ribosomal genes occurs via distinct pathways for ribosomal protein genes and ribosomal biogenesis genes, which can be separated based on genetic requirements for repression and based on chromatin changes during gene repression. Together, our dynamic studies provide a rich resource for investigating chromatin regulation, and identify a significant role for the "activating" mark H3K4me3 in gene repression.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Comment in
-
Chromatin proteins: key responders to stress.PLoS Biol. 2012;10(7):e1001371. doi: 10.1371/journal.pbio.1001371. Epub 2012 Jul 31. PLoS Biol. 2012. PMID: 22859908 Free PMC article.
References
-
- Kornberg RD, Lorch Y (1999) Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell 98: 285–294. - PubMed
-
- Clapier CR, Cairns BR (2009) The biology of chromatin remodeling complexes. Annu Rev Biochem 78: 273–304. - PubMed
-
- Kouzarides T (2007) Chromatin modifications and their function. Cell 128: 693–705. - PubMed
-
- Strahl BD, Allis CD (2000) The language of covalent histone modifications. Nature 403: 41–45. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
