

Group normalization for genomic data
- PMID: 22912661
- PMCID: PMC3418286
- DOI: 10.1371/journal.pone.0038695
Group normalization for genomic data
Abstract
Data normalization is a crucial preliminary step in analyzing genomic datasets. The goal of normalization is to remove global variation to make readings across different experiments comparable. In addition, most genomic loci have non-uniform sensitivity to any given assay because of variation in local sequence properties. In microarray experiments, this non-uniform sensitivity is due to different DNA hybridization and cross-hybridization efficiencies, known as the probe effect. In this paper we introduce a new scheme, called Group Normalization (GN), to remove both global and local biases in one integrated step, whereby we determine the normalized probe signal by finding a set of reference probes with similar responses. Compared to conventional normalization methods such as Quantile normalization and physically motivated probe effect models, our proposed method is general in the sense that it does not require the assumption that the underlying signal distribution be identical for the treatment and control, and is flexible enough to correct for nonlinear and higher order probe effects. The Group Normalization algorithm is computationally efficient and easy to implement. We also describe a variant of the Group Normalization algorithm, called Cross Normalization, which efficiently amplifies biologically relevant differences between any two genomic datasets.
Conflict of interest statement
Figures





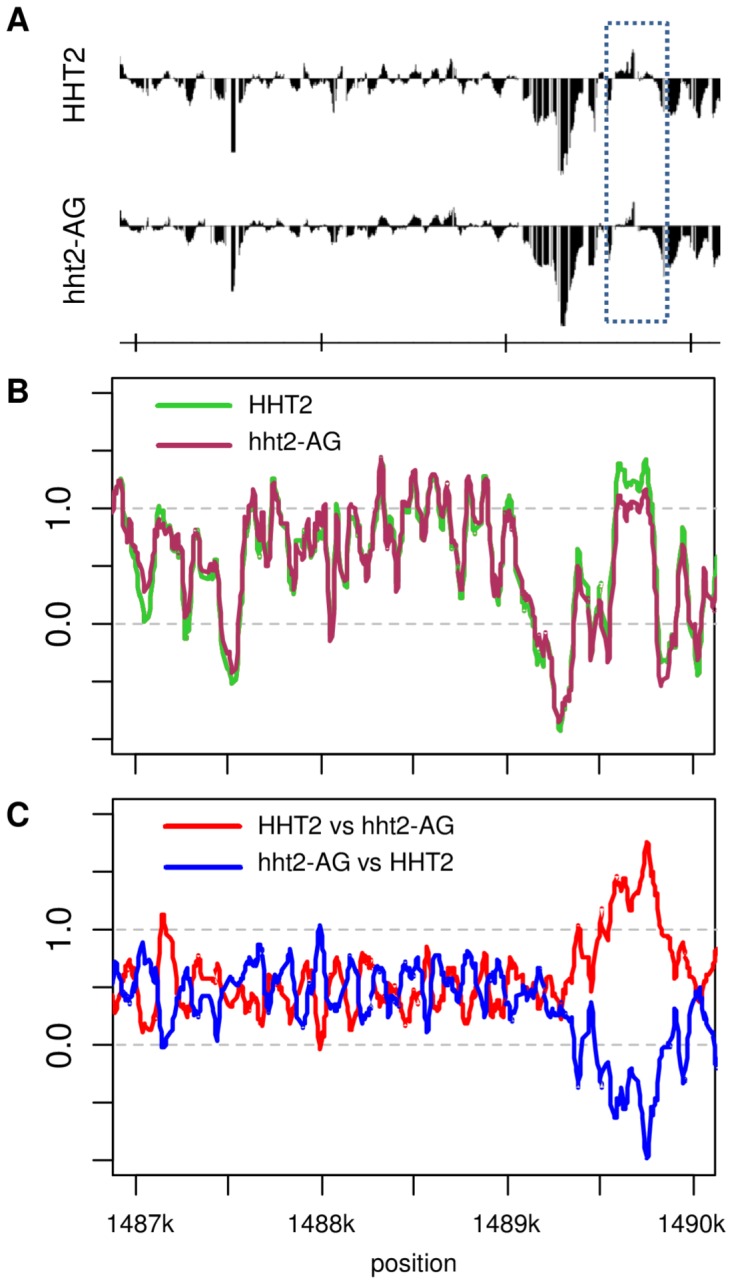
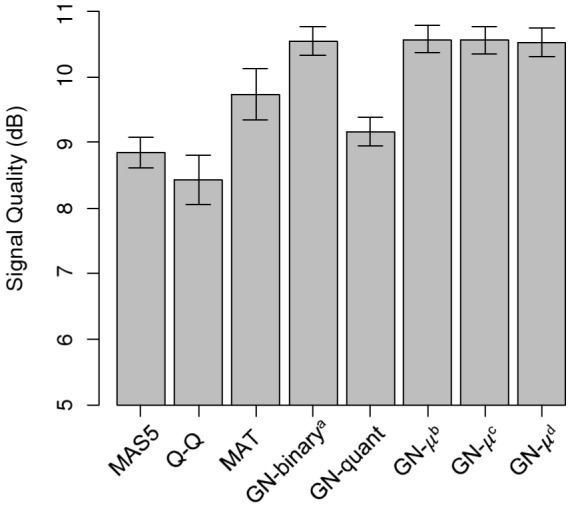

Similar articles
-
Smooth quantile normalization.Biostatistics. 2018 Apr 1;19(2):185-198. doi: 10.1093/biostatistics/kxx028. Biostatistics. 2018. PMID: 29036413 Free PMC article.
-
CGHScan: finding variable regions using high-density microarray comparative genomic hybridization data.BMC Genomics. 2006 Apr 25;7:91. doi: 10.1186/1471-2164-7-91. BMC Genomics. 2006. PMID: 16638145 Free PMC article.
-
Delineation of amplification, hybridization and location effects in microarray data yields better-quality normalization.BMC Bioinformatics. 2010 Mar 26;11:156. doi: 10.1186/1471-2105-11-156. BMC Bioinformatics. 2010. PMID: 20346103 Free PMC article.
-
A stepwise framework for the normalization of array CGH data.BMC Bioinformatics. 2005 Nov 18;6:274. doi: 10.1186/1471-2105-6-274. BMC Bioinformatics. 2005. PMID: 16297240 Free PMC article.
-
Using generalized procrustes analysis (GPA) for normalization of cDNA microarray data.BMC Bioinformatics. 2008 Jan 16;9:25. doi: 10.1186/1471-2105-9-25. BMC Bioinformatics. 2008. PMID: 18199333 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
